Process NMR spectrometry
Alison Nordon, Colin A. McGill and David Littlejohn*
Department of Pure and Applied Chemistry/CPACT, University of Strathclyde, 295 Cathedral Street, Glasgow, UK G1 1XL. E-mail: d.littlejohn@strath.ac.uk
First published on 25th January 2001
 Alison Nordon Alison Nordon | Alison Nordon is a postdoctoral research assistant with the Centre for Process Analytics and Control Technology (CPACT) at the University of Strathclyde. She obtained both her BSc (Hons) degree in chemistry and PhD degree in solid-state NMR from the University of Durham. Alison’s current research interests include the development of low-field NMR and acoustic spectrometries for use in process analysis, and data analysis methods for spectroscopic techniques. |
 Colin A. McGill Colin A. McGill | Colin McGill is a postdoctoral researcher at the Centre for Process Analytics and Control Technology (CPACT), University of Strathclyde. He completed his PhD studies at the University of Strathclyde, in process analytical chemistry. Colin graduated from the University of Strathclyde in 1996 with first class honours in Forensic and Analytical Chemistry. He then took up a position with Merck Ltd., before returning to carry out his PhD studies. His current research interests are in developing spectroscopic techniques for the on-line monitoring and control of chemical processes. |
 David Littlejohn David Littlejohn | David Littlejohn has been Professor of Analytical Chemistry at the University of Strathclyde since 1988. He has broad interests in analytical science and has published more than 140 papers on a range of topics. In 1997, he was one of the co-founders of the Centre for Process Analytics and Control Technology (CPACT), which is a multi-partner, multi-disciplinary research collaboration between industry and academia. |
1 Introduction
NMR spectrometry is a widely used analytical technique for structural elucidation and identification of chemical species in the analysis of many different materials including organic chemicals, inorganic complexes and large biological molecules. In this context, the instrumentation used comprises a high-field superconducting magnet (typically 200–750 MHz), and so detailed chemical shift and coupling-constant information may be extracted from the high-resolution spectra obtained. At the moment NMR spectrometry is not commonly used in process analytical chemistry. Indeed, NMR spectrometry has been mentioned for the first time in the most recent1 of a number of reviews of process analytical chemistry,2,3 but only then within a miscellaneous techniques section. This is partly due to NMR techniques being rather complicated and the signals obtained can be difficult for non-NMR specialists to understand.4 NMR instruments can be expensive and the technique is much less sensitive than others, e.g., IR spectrometry.However, NMR spectrometry has the potential of being a very useful technique in process environments as it is non-destructive and does not require the measurement probe to be inserted into the process liquors, which avoids fouling.4 Another problem that was faced when NMR spectrometry was initially applied in at-line or on-line process analysis was that high-field laboratory instruments were simply moved into the plant. Process operators found problems in the calibration and maintenance of these instruments. Recently, small, dedicated low-field (<60 MHz) NMR systems, based on permanent magnet technology, have been developed, which in turn has led to more reports of at-line and on-line process applications.5–7
As interest in the use of NMR spectrometry in process applications is increasing, this review of the subject has been prepared. The report covers various aspects of the technique that are relevant to NMR analysis of static or flowing materials (e.g., foods, chemicals and polymers). The review begins with a discussion of the factors that affect NMR measurements of flowing samples. This is followed by sections on low-field NMR applications, on-line process applications and the use of magnetic resonance imaging in industry. Brief details are given on current commercial instrumentation. The final section describes chemometric and data analysis procedures used to acquire chemical and process information from NMR measurements, especially when overlap of resonances occur, as is often encountered in low-field NMR spectra.
2 General aspects of NMR measurements of flowing materials
When a sample is placed in a static magnetic field, B0, the nuclear spins precess about B0 at the Larmor frequency. The spins align themselves with the magnetic field resulting in a net magnetisation, M0, parallel to B0 (z-axis). Assuming exponential behaviour, the rate at which M0 builds up along B0 is given by 1/T1, where T1 is the spin-lattice or longitudinal relaxation time. Upon application of a rf field, typically for a few microseconds, this rotates M0 away from the z-axis into the xy plane. The rate at which the spins relax to give no preferred orientation in the transverse plane is given by 1/T2 where T2 is the spin–spin or transverse relaxation time. T1 is not only a measure of the time required to create M0 initially, i.e., upon placement of the sample in B0, but it also describes the time for the magnetisation to return to equilibrium after application of a rf field.Both relaxation times affect the signal obtained in a NMR experiment. The linewidth of the absorption signal (after Fourier transformation) is given by 1/T2* where T2* is the effective T2 in the presence of, for example, magnetic field inhomogeneities. T2* is effectively the time required for the signal in the time domain (FID) to decay to zero. Commonly in NMR spectrometry, when the signal-to-noise ratio is not good, the FID signal is multiplied by an exponentially decaying function in order to ‘remove’ the noise at longer acquisition times. This post-acquisition treatment of the data also increases the rate at which the signal decays, thus increasing the line width of the signal; hence this type of data treatment is referred to as line broadening. The area, or integral, of the NMR signal is given by the magnitude of M0 and thus is dependent upon the time that the sample is in the magnetic field, the recycle delay (i.e., the delay between pulses) and T1.
To perform quantitative NMR measurements, it is necessary to use a recycle delay of at least 5 times the longest T1 (≈99% signal relaxation) in the sample to eliminate any possible saturation and nuclear Overhauser enhancement8 (NOE) effects. The latter effect can actually reduce the signal area, despite its name, particularly for large molecules and for those nuclei that possess a negative gyromagnetic ratio,8e.g., 29Si and 15N. For the study of nuclei with long T1 times (e.g., 13C), quantitative experiments are extremely time consuming due to the considerable recycle delay needed and the low natural abundance of the 13C nucleus. Indeed, in such cases, it is difficult to obtain simply a good signal-to-noise spectrum, let alone achieve quantitative results (at least for every nucleus in the sample). However, it is possible to optimise the pulse flip angle and recycle delay9 to obtain the best signal-to-noise possible in the least amount of time. A major drawback of this approach is that it is not compatible with more complicated pulse sequences. In order to reduce experimental time, yet still enable data to be interpreted quantitatively, relaxation agents such as chromium triacetylacetonate Cr(acac)3 may be added to the sample solution. However, there are several disadvantages to this procedure, which include the obvious contamination of the sample of interest, perturbation of chemical shift and line shape, and limited solubility of Cr(acac)3 in the sample solution.10
For a flowing system, the effects of the nuclear spins ‘moving’ through B0 and the rf field must be taken into account. If it is assumed that the spins are completely polarised, i.e., sufficient time has been allowed for M0 to build up, the observed relaxation times for a flowing system can be given by,
 | (1) |
The observation of an increase in signal area with an increase in flow rate implies that the spins must be completely polarised prior to reaching the rf coil. However, if the effects from incomplete polarisation are included,14,15 then the variation of signal area with flow rate can be divided into two distinct flow regimes. The division of flow into slow and fast regimes is based on the residence time of the sample in the detection region (τ) and the time allowed for polarisation of the spins (Tp) with respect to the recycle delay (Td) used in the experiment.4 It will be assumed in the following discussion that Td is less than 5 × T1 and hence saturation of the nuclear spins occurs in the static experiment. At low flow rates, where Tp is greater than 5 × T1, an increase in signal area is observed as the flow rate is increased due to the factors discussed above. However, once the rate of replenishment of saturated with unsaturated spins becomes comparable to the relaxation rate, the saturation effects are removed. Hence, any further increase in flow rate will yield the same signal area. At high flow rates, where Tp is less than 5 × T1, a decrease in signal area is observed as the flow rate is increased due to incomplete polarisation of the nuclear spins prior to reaching the detection coil. Hence, in a plot of signal area against flow rate (Fig. 1) a maximum will be observed at intermediate flow rates due to the combined effects of saturation and incomplete polarisation of the spins. Such a variation in signal area with flow rate has major implications for the attainment of quantitative data.16–18
 | ||
| Fig. 1 Variation in the area of a NMR peak as a function of flow rate. | ||
Earlier it was mentioned that the relaxation time for a flowing system is less than that for a static system. Hence, flow NMR can be used to reduce T1 for the sample and enable a shorter recycle delay to be employed, as has been demonstrated for 13C, 31P, 113Cd, 29Si and 15N.10,19,20 In many studies, a so-called premagnetisation coil has been incorporated into the probe design21 (see later) in order to ensure complete polarisation of the sample prior to entering the rf coil. If a sufficient recycle delay is also used, so as to eliminate saturation effects, flow NMR data can be interpreted quantitatively. For example, 29Si and 13C NMR experiments have been performed on flowing systems to determine the molecular weight of siloxane polymers.10,16 The conditions required for quantitative flow NMR measurements have been investigated particularly with respect to HPLC-NMR22,23 experiments (see later).
Flow NMR has also been utilised in dynamic nuclear polarisation (DNP) experiments to enhance the signal-to-noise ratio.24 A potential disadvantage of flow NMR is that, for those nuclei which exhibit a positive NOE enhancement, the gain in sensitivity from this source can be lost. On the other hand, for those nuclei which exhibit a negative NOE enhancement, flow NMR can remove the decrease in signal area. However, NOE enhancements have been observed in continuous flow 13C (positive NOE) and 15N (negative NOE) NMR measurements using upstream 1H pre-irradiation of the sample.25
An alternative method to flow NMR for replenishing saturated spins involves moving the sample up and down (parallel to the magnetic field) in a custom-built probe.26 In this procedure, the sample was locked into position during excitation and data acquisition, and was then quickly moved to a new position by a stepping motor. Sampling was interlaced so that different parts of the sample were sampled during the +z translation part of the cycle to those sampled during the reverse −z part.
In many studies, no allowance was made for the flow characteristics of the liquid, which affect the expressions describing the signal area and width.27 Types of flow include laminar, turbulent and plug flow. In laminar flow, which tends to occur at low flow rates, the liquid velocity varies with distance from the tube axis. Therefore, volume elements of the liquid entering the detector coil will have been in the field for times proportional to the distance from the tube axis. In turbulent flow, the velocity pattern changes randomly and velocity characteristics cannot be defined with precision, but its statistical features such as mean velocity, velocity variance and time-averaged velocity profile are all well defined. Equations simulating the steady-state magnetisation of liquids in continuous-flow FTNMR have been derived using a classical vector model, assuming plug flow,28 from which the conditions for the attainment of the optimum signal-to-noise ratio have been deduced.
NMR spectrometry is an inherently insensitive technique and thus to obtain the best signal-to-noise ratio large detection volumes are favourable. However, in HPLC-NMR spectrometry large sample volumes are unfavourable as the separation ability of the HPLC column degrades with an increase in injection volume.29 Therefore, in the design of probes for HPLC-NMR studies, an ‘intermediate’ detection volume has been used. The signal-to-noise (S/N) ratio of a NMR detection cell can be defined by30
 | (2) |
Earlier it was mentioned that many probe designs incorporate some sort of premagnetisation region. This enables the spins to be completely polarised prior to reaching the detection region and so a larger signal can be obtained. In the two examples shown in Fig. 2, one taken from a flow NMR experiment21 and the other from a HPLC-NMR design,13 the rf coil is positioned in the centre of a polarising coil. This set-up enables the sample to be present in the magnetic field for a longer time prior to entering the detection coil. Zhernovoi and Latyshev employed an additional powerful polarising magnet 100 cm in advance of the detector magnet.15 Since the decay of magnetisation during the passage along the pipe connecting the two magnets was small for the pure water sample used, the magnetisation of polarised nuclei entering the second magnet was greater than it would have been without the first magnet. Hence, this arrangement can be used to give an increased signal area and ensure complete polarisation of the spins prior to reaching the rf coil, as long as the distance between the two magnets is not on the same timescale as the T1 relaxation.
 | ||
| Fig. 2 NMR probes incorporating a premagnetisation region. (a) Reprinted from M.C. McIvor, J. Phys. E, 1969, 2, 292 by courtesy of Institute of Physics Publishing Limited. (b) Reprinted with permission from J. F. Haw, T. E. Glass, D. W. Hausler, E. Motell and H. C. Dorn, Anal. Chem., 1980, 52, 1135. Copyright 1980 American Chemical Society. | ||
Although flow NMR has been successfully used in 1H, 19F and 13C experiments to enhance the signal obtained, the major area of application of flow NMR is in HPLC-NMR spectrometry.33,34 HPLC-1H NMR has been used extensively in the pharmaceutical industry.35 The major advantage of this technique, for example in the study of drug metabolites in fluids,36 is that it permits rapid and unambiguous detection of such species without any pretreatment of the sample. In studies of polymers, HPLC-1H NMR measurements have been used to determine the degree of polymerisation in polyethylene oxide37 (PEO) and the tacticity of oligostyrenes.38 It has also been possible to determine the average degree of substitution of aromatic ring systems in fuels.31 HPLC-1H NMR spectrometry has also been used to analyse ground water samples collected from sites situated near former ammunition plants.39 Without special optimisation of the HPLC method, it was possible to detect substances present on the microgram per litre scale.
Early HPLC-NMR experiments were either stopped-flow40 or performed using very low flow41 rates in order to improve the signal-to-noise ratio. However, continuous flow is now far more favourable primarily due to the advances in magnet technology, which have resulted in higher B0 values [S/N ∝ B03/2, as indicated in eqn. (2)]. Continuous flow measurements are more favourable than stopped-flow, not only due to the shorter recycle delay that can be used in the former and the subsequent saving in time that this represents, but also because sample heterogeneities are less of a problem.4 One of the major problems in HPLC-1H NMR spectrometry is the signal that arises from the solvent and the requirement for the receiver to be able to digitise over a large dynamic range. It is possible to use deuterated solvents to remove this problem, although the use of capillary HPLC-NMR spectrometry for stopped-flow experiments has been reported, which requires a smaller sample volume.42
Solvent presaturation schemes for single detection coil continuous flow probes are unfavourable as presaturated nuclei are being continuously removed from the flow cell. Other methods for removing the solvent peak from the spectrum, in the case of flowing systems,43 include selective excitation, binomial solvent suppression sequences44 and post-acquisition solvent signal removal.45–47 An alternative to using deuterated solvents, is the use of supercritical fluids (SFC), but this requires modification of the NMR probe.48–50 Other disadvantages of SFC-NMR spectrometry include the pressure dependence of the chemical shifts and also an increase in the T1 of the system (compared to that in the liquid state), which increases the experiment time. If the analyte compounds contain fluorine then HPLC-19F NMR spectrometry may be a viable alternative to HPLC-1H NMR spectrometry. The major advantage of 19F NMR, in comparison to 1H NMR, is that, for the most commonly used solvents, there will be no large solvent peak present in the sample spectrum.22,51 HPLC-19F NMR spectrometry has been used to examine mixtures of p-fluorobenzoate derivatives of simple alcohols and phenols,51 and has found application in the pharmaceutical industry where many drugs contain fluorine.
Non-chromatographic applications of flow NMR spectrometry include the study of flowing systems and reaction kinetics. NMR can be used to determine flow rates and study other related phenomena such as velocity distributions and profiles.52 Time-averaged NMR imaging techniques (see later section) have been used to determine velocity profiles and turbulence intensities.53,54 Flow NMR spectrometry has been utilised in the study of reactions occurring on the 50 ms to 5 min timescale and also in the identification of transient intermediate species.55,56 NMR spectrometry is a much more powerful diagnostic tool than UV–visible spectrometry, which is commonly used to monitor reactions. NMR can be used to identify the species present and thus can aid in the assignment of UV-visible spectra. For example, with continuous flow 1H NMR it was possible to identify a thermodynamically unstable complex formed in the reaction of 3,5-dinitrocyanobenzene with methoxide ions.55 In the reaction of 2,4,6-trinitrotoluene and methoxide ions,55 it was possible to identify the rapid formation of one species alongside the slower formation of another. From the UV-visible spectrum it had only been possible to postulate that a number of species might exist. In the reaction of thiamine with hydroxide ions,57 it was possible to follow the slow conversion of the yellow thiol form to the colourless thiol form; previously it had only been possible to detect a mixture of the two species.
A high-pressure 1H NMR probe (stopped-flow) has been designed to monitor fast reactions at pressures up to 200 MPa.58 Using this equipment, it has been possible to measure activation volumes for reactions with half-lives longer than a few seconds, which could not be followed spectrophotometrically on the same timescale. A high-pressure flow cell has been designed to enable homogeneous catalysis to be studied in situ at pressures and temperatures in the ranges of 1 to 200 bar and −40 to 175 °C, respectively.59 The probe design ensures good mixing between the gas (CO) and the solution, and also maintains the concentration of gas in solution. Flow probe technology has also been used in flow injection NMR experiments which permit rapid analysis of a large number of samples.60 Stopped-flow photo-CIDNP (chemically induced dynamic nuclear polarisation) has been used to observe protein folding.61 In solid-state NMR, probes have been designed which permit the study (in situ) of hydrocarbon conversions on solid catalytic surfaces, and the adsorption and desorption of 13C-labelled benzene on a catalyst bed.62,63
Probes employing microcoils have been designed to permit analysis of smaller sample volumes with an improved signal-to-noise ratio in comparison to that obtained using conventional rf coils.64 For example, through the use of these coils, detection limits of less than 50 ng were possible for amino acids, with an analysis time of 1 min.65 These small volume coils have enabled on-line NMR detection for capillary electrophoresis65,66 and microscale separations by HPLC.67
In order to increase the throughput of samples, probes have been designed that incorporate multiple microcoils. In one approach, termed multiplex NMR,68,69 four coils were connected in parallel and through the use of gradients, signals could be detected simultaneously (with a single receiver) from four samples. Simultaneous acquisition of NMR data from multiple samples has also been demonstrated using a probe incorporating two sample coils, two duplexer/preamplifier stages and two receivers.70 An alternative method that permits analysis of multiple samples using multiple coils, separates the individual signals by careful timing of data acquisition in recycle delays.71 Although all the above examples involved the analysis of static samples, after suitable development it is envisaged that this method could be used in applications such as process monitoring.68 Recently, a NMR flow probe has been described for use in direct injection sample analysis.72 The sample is loaded into the flow cell through the bottom inlet of the probe, via a low diameter probe that can be connected to a liquids-handling robot. Retrieval of the sample is also via the bottom inlet, which was found to reduce the number of times that the flow cell needed to be rinsed between samples.
3 Applications of low-field NMR spectrometry for analysis of static process samples
Most low-field NMR applications involve the measurement of spin–spin (T2) and/or spin–lattice (T1) relaxation times, which are then related to a specific physical property such as viscosity, surface area and moisture content. Other applications involve analysis of the FID or spin–echo to yield quantitative information relating to the concentrations of individual components, which can be distinguished by virtue of different T2 values.73 In this context, the data obtained by low-field NMR is low resolution.Low-field, low-resolution NMR spectrometry has been used extensively in a variety of industries including those involving the manufacture of food, pharmaceuticals and petrochemicals. Example applications include the determination of moisture content,74,75 fat content,76,77 hydrogen content and fluorine content.78,79
One of the most common areas of application of low-field, low-resolution NMR spectrometry has been the determination of water in samples. This includes the quantitative determination of moisture content in, for example, corn,80 zeolites81 and soil.82–84 For most analyses, a low-field NMR spectrometer was used with an operating frequency of 20 MHz for 1H. However, in the analysis of soil, single-sided magnet designs were used to enable the instruments to be deployed in the field. In one example, a 3 MHz NMR spectrometer was mounted on the back of a tractor.83 Another area of interest includes investigation of the state of water in samples. For example, a number of studies have looked at the variation in 1H relaxation times of gelatin gels as a function of water content.85–87 An increase in T2(1H) was observed for gels containing 15–20% moisture. Maxima were also observed in plots of T1(1H) and cross relaxation rates against moisture content at a moisture content of 15%. These observations were interpreted as the formation of a monolayer of water on the gel at moisture levels of 15%, followed by the formation of multilayers of water between 15% and 20% water. Water droplet size distributions in solid and liquid emulsions have been determined using pulsed field gradient (PFG) NMR spectrometry.88,89 A 4.1 MHz NMR spectrometer was used to determine the moisture content of samples of rice, which were contained within 40 mm NMR tubes.90
Drying processes have been analysed using low-field, low-resolution NMR spectrometry. For example, a solid-echo pulse sequence was used to monitor the drying of grated and cubed carrots, with an analysis time of less than 1 min.91 The binding of water to milk proteins92 and powdered milk93 has been studied using T2 relaxation times made on a 20 MHz NMR spectrometer. As the exchange rate between water molecules bound to the solid matrix and ‘free’ bulk molecules was slow on the NMR timescale, non-exponential spin–echo decay curves were obtained. Thus it was possible to determine the proportion of water that was bound to the milk product surface. The oil and water content in emulsions has been determined using T1 relaxation times on a low-field NMR spectrometer.94
It has been shown that low-resolution NMR (20 MHz) can be used for the determination of ethanol in alcoholic beverages.95 It was possible to separate the 1H NMR signal from water and the ethanol OH from that of the ethanol CH2 and CH3 groups using the indirect, or J, coupling constant. When the refocusing time, or τ, in the spin–echo sequence was set to 68 ms, a signal was only obtained from the water and ethanol OH. In comparison, with a standard Carr–Purcell–Meiboom–Gill (CPMG) sequence, the signal obtained was from all the protons present in the sample. Hence, it is possible to determine the ethanol content in the sample. The presence of sugars was found to have no effect on the signal obtained. However, the accuracy obtained (±0.5% v/v over the range 0 to 70% v/v) was found to be insufficient for assays of final products of wines, beers and spirits.
Alcohols are added to gasolines as fuel extenders and to improve the octane rating.96 They can also be added by crooked fuel dealers as dilutants and the gasoline falsely sold as pure. Low-field, high-resolution 1H NMR spectrometry at 60 MHz has been used to monitor alcohols in fuels. There are no signals from the gasoline in the 2.8–6.8 ppm spectral region. Hence, it is possible to observe the methanol CH3 singlet at 3.4 ppm. The peak from the methyl protons of methanol was integrated for various concentrations of methanol and a linear plot of peak area versus percent volume of methanol was obtained between 0–25% v/v, with a detection limit of approximately 0.06% v/v. Seventy samples were analysed using this approach, and twelve samples were found to contain methanol at concentrations between 3 and 10%. Methanol is not the only alcohol used as an additive/adulterant in gasolines. It was possible to identify the presence of other alcohols from the 1H NMR spectrum.
The spin–lattice relaxation time, T1, can be used to determine the surface area of a sample. Relaxation times were determined, at 20 MHz, for a number of different fluids, including water, methanol, ethanol and cyclohexane, on samples with different surface areas.97 The surface area of the sample was found to be inversely proportional to the spin–lattice relaxation time of the liquid on the surface.
For the measurement of the fat content of cracker biscuits,98 the CPMG pulse sequence was used on a 20 MHz spectrometer. The 1H NMR signal obtained was proportional to the fat content of the crackers when the moisture content was less than 7.1%. Above this value, the water was found to make a contribution to the signal obtained. The mobility of lipids in low-moisture bread has been investigated using low-field 1H NMR experiments at 20 MHz.99 This was done using a combination of relaxation time data, i.e., T1 and T2, and diffusion coefficients, D, which were determined using pulsed field gradient experiments. It was decided that the lipids are distributed in globules dispersed in the bread matrix and diffusion of the lipids occurs within these globules.
A variety of dairy products, e.g., whipping cream, whole milk and skimmed milk, has been analysed using 1H NMR spectrometry at 42 MHz.5 Although spectra were dominated by the water signal, it was possible to see the fat signal at 1.3 ppm, which was particularly evident in the spectrum of whipping cream. Samples were contained within 5 mm NMR tubes, which were spun at 20 Hz during analysis.
An external unit has been developed to allow low-frequency measurements, in the 700 kHz to 1 MHz range, to be made using a commercial high-field NMR spectrometer.100 The application of such an instrument, at 700 kHz, was demonstrated using hen eggs shells, where the relaxation time measurements can give an indication of the freshness of the egg.
NMR relaxation time measurements have been used to study two key processing steps in the dairy industry; acidification and syneresis.101 For example, for samples with a pH above 5.3, T2 was found to be correlated with the amount of solubilised micellar calcium phosphate. During the syneresis process, the relaxation time parameters can be used to quantify, non-invasively, the amount of whey expelled and the curd humidity.
A 20 MHz spectrometer has been used for viscosity measurements of bitumens.102 Bitumens consist of three phases; semi-liquid, solid-like mobile and solid-like rigid. Each phase has a different relaxation time (semi-liquid with the longest T2 time). If the exponential decays of each phase are extrapolated back to zero time (i.e., when the pulse was applied) the magnetisation due to each particular phase is obtained. Division by the total magnetisation gives the proportion of each phase in the sample. T2 measurements were made for different bitumens of known viscosity and the results were found to correlate well. As would be expected, higher viscosity values were obtained for bitumens with a high content of solid-like rigid phase material.
The extent of cure in an epoxy resin has been determined using the shape and intensity of the FID.103 Transverse relaxation times and the FID signal intensity at specific points have been used to monitor gel formation.104,105
4 On-line applications of process NMR spectrometry
There have been several reports of NMR measurements of flowing systems in process analysis. A common application of process NMR is the determination of moisture content.4,7,73,106–109The moisture content of coal, which was fed through the sample coil from a hopper under gravity at a rate of 100 g per hour, has been determined.106 The analyser, which operated at 16 MHz, measured water content over the range of 0–14% with an accuracy of ±1% moisture. The spectrum produced from the measurements was the first derivative of the absorption signal. The peak-to-peak amplitude of this was found to correlate to the amount of moisture present. The moisture content of coal samples flowing through a 3/8 inch pipe has been determined using a 30 MHz NMR spectrometer.107 After removal of water from the samples by drying, the 1H NMR signal intensity was used to determine the hydrogen content of the coal.
The analysis of moisture in grain samples has been the subject of a number of studies.7,108,110–112 In one study, a sample of moist wheat was transported to the probe of a 20 MHz process NMR spectrometer.7 The proton signal from water and hydroxyl groups diminishes at a slower rate than the signal arising from protons chemically bound to the carbon in wheat, thus giving the free induction decay (FID) two parts; an initial fast decay followed by a slower decay. By regressing the two components of the FID to the y-axis, the ratio of hydrogen from water to bound hydrogen was obtained. It was found that the amount of bound hydrogen was approximately proportional to the amount of wheat in the detector coil. Hence, the ratio of the different protons gave the moisture content of the wheat without the sample having to be weighed. The results obtained were then used as an input to a control valve, which adjusted the flow of water to the wheat. For corn and oilseeds it was observed that the protons from the oil present were also contributing to the signal from the moisture. Hence, a Hahn spin–echo sequence was used to refocus the oil signal, and to remove the water signal. This gave a signal whose intensity is proportional to that of the oil. By subtracting this value from the intensity obtained for the unbound protons, the moisture content was obtained.
A study of cereal products was carried out by Nicholls and De Los Santos.108 They decided to develop a moisture sensor for the wet corn milling industry after a study by the American government found that drying processes in industry consumed approximately 8% of the total industrial energy used in the USA. Proton NMR spectrometry was chosen to be developed as a moisture sensor for many reasons, including its applicability to many processes including the food, tobacco, textile, agricultural products and paper industries. The sensor can be configured in many ways, e.g., to analyse materials flowing in a pipe, to measure materials on belts or chutes or even paper coming off presses. Using NMR spectrometry, the moisture content is measured throughout the material so there are no problems encountered from inhomogeneous water distribution. Another major advantage described is the fact that there are no moving parts in the analyser and no contact between the probe and the sample. The instrument used operated at 11 MHz. The sampling system (shown in Fig. 3) was a hole in the chute which allowed the corn to fall into the probe. After measurement, a piston expelled the sample back into the process stream. This piston also contained cavities which contained calibration materials. This allowed the analyser to be calibrated before each measurement. Two different measurements were taken to evaluate the procedure. The first was the FID ratio method, as used by Pearson et al.7 This allowed the fast measurement of up to 30% moisture. Secondly, a CPMG sequence was used. This pulse sequence required more computer processing and took longer to acquire results. The advantage of this sequence was that moisture contents of up to 60% could be determined.
 | ||
| Fig. 3 Sampling system for moisture content measurements. Reprinted from C. I. Nicholls and A. De Los Santos, Drying Technol., 1991, 9, 849 by courtesy of Marcel Dekker Inc. | ||
Tellier et al.109 carried out measurements of lipids and water content in a flow of fine meat paste using a 90 MHz NMR spectrometer. Using a flow rate of 7.5 cm s−1 the repeatability of the measurements was found to be ±1%, which allowed detection in the variation of water or lipids of 3%, with a measurement time of less than 1 min.
A small on-line process NMR spectrometer has been used to monitor the moisture content and surface area of reduction-grade aluminium oxide as it leaves a rotary kiln.113–115T1 measurements at 20 MHz were made on-line and also in stopped-flow mode, for aqueous slurries of titania and glass.116 From the results obtained it was possible to determine the solids content of the slurries and also the surface area of the solids. Viscosity measurements on polymers have been carried out on a 60 MHz spectrometer, using a stop-flow technique.117
There are only a few reports of NMR spectrometry being used to monitor the chemical composition of a process stream. As early as 1956, a process analyser was designed to monitor the ratio of two organic liquids flowing through the probe of a 30 MHz continuous wave spectrometer.118 Haw and Skloss5,119 have designed a prototype flow NMR system where two HPLC pumps were used to pump hexane and toluene, which were then mixed and passed through the probe of the spectrometer. The instrument had an operating frequency of 42 MHz for 1H, and employed a lock channel and automated shims. The amount of toluene in hexane was monitored by integrating the aromatic proton signal at 7.1 ppm and the methyl proton signal at 2.3 ppm. Other sample mixtures that were analysed include sucrose in water and ethanol in water.5 In all cases, a flow rate of 2 mL min−1 was employed.
The same authors reported on the use of proton NMR to determine the concentration of oxygenates in fuel.6 A 42 MHz spectrometer was employed and the samples flowed through the probe at a rate of 2 mL min−1. Fig. 4 shows the 1H NMR spectra obtained of different concentrations of methyl tert-butyl ether (MTBE) in gasoline. Due to environmental constraints, fuel sold in many parts of the USA must contain 2.0–4.0% w/w of oxygenates to make the combustion process cleaner. The low-field NMR method had a measurement time of approximately 60 s compared with 20 min for gas chromatography. It was found that a gasoline sample has no NMR peaks in the region of 3.0–4.6 ppm, which is where the proton peaks of the oxygenates exist. Therefore, the concentration of oxygenates in the sample can be measured with no interference. The method was found to work well when only one or two different oxygenates were blended into the fuel. Problems arose when three or four different oxygenates were added to the same gasoline.
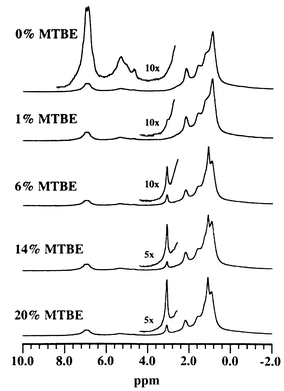 | ||
| Fig. 4 1H NMR spectra obtained at 42 MHz with a flow rate of 2 mL min−1 of different concentrations of methyl tert-butyl ether (MTBE) in gasoline. Reprinted with permission from T. W. Skloss, A. J. Kim and J. F. Haw, Anal. Chem., 1994, 66, 536. Copyright 1994 American Chemical Society. | ||
Determination of the concentration of H2SO4 present in an alkylation process has been carried out using on-line 1H NMR spectrometry.120 It was possible to determine the acid concentration (percent weight) from the chemical shift difference between the acid peak and the hydrocarbon peaks.
In cases where the components of interest give rise to similar spectra, derivatisation may aid quantification of the components of interest. For example, it is difficult to determine the ratio of primary to secondary hydroxyl groups in a mixture of polyethylene and polypropylene glycols (PEG and PPG, respectively) from 1H NMR spectra acquired at 57 MHz.119 Unfortunately, a number of problems were encountered in the on-line derivatisation of polyol mixtures using trifluoroacetic acid (TFA). However, the principle of on-line derivatisation followed by analysis of the resultant mixture by on-line 19F NMR spectrometry was demonstrated using some simpler alcohols.
A stopped-flow procedure121 has been used to monitor the last step of the synthesis of retinoic acid, which is a Wittig reaction. 31P measurements were acquired for the monitoring of reactants and products. As the reaction proceeds, the 31P signal from the ylide at 13.5 ppm (referenced to H3PO4) diminishes and is replaced by the signal at 25.5 ppm from triphenylphosphine oxide. The samples were pumped from a 0.5 L laboratory reactor into a 10 mm sample tube in a Bruker WM360 spectrometer operating at 360 MHz for 1H. After measurement, the sample was pumped out and replaced by a fresh sample. The total measurement time for each spectrum was 135 s.
The use of proton NMR spectrometry has been investigated for the characterisation of liquors used in the pulp and paper industry.122 Experiments were conducted at both 60 and 400 MHz. The magnitude of the FID obtained from black liquor samples was found to have a linear relationship with the percentage of solids in the liquors. The analysis was carried out on static samples, although the authors believe that the results could be reproduced in a flowing system. T1 and T2 measurements were also made for each of the liquors of known viscosity, but with different % solids contents, to see whether there was a correlation between relaxation time and viscosity. Unfortunately, it was not possible to establish a general relationship between viscosity and relaxation times for the range of samples investigated.
5 Industrial applications of magnetic resonance imaging (MRI)
When a sample is placed in a static magnetic field, B0, the nuclear spin states of those nuclei with I > 0 (I is the nuclear spin quantum number) become non-degenerate. Upon application of a rf field (of the appropriate frequency), transitions can be induced between the various nuclear spin states. Hence, it is possible to distinguish between nuclei of different elements and also nuclei of the same element but in different chemical environments by virtue of their resonance frequencies. In magnetic resonance imaging (MRI) an additional small magnetic field gradient is applied such that the resonance frequency of the nuclei varies as a function of their position in real space. In the simplest case, the gradient is applied parallel to the static magnetic field, i.e., along the z direction, to yield a 1D spectrum. However, by application of gradients along the x, y and z directions, it is possible to obtain a 3D image of the sample of interest. Perhaps the most widespread use of MRI is as a diagnostic tool in the field of medicine. However, over recent years the potential of MRI techniques in the process industries has been recognised.123–126MRI has been used to monitor polymerisation reactions and to determine structural characteristics of polymers.126,127 An example of a polymerisation reaction studied is that of methyl methacrylate in which MRI was able to monitor the disappearance of the monomer as the reaction proceeded.128 The information obtained from such an experiment could be used to reveal any differences in polymerisation rate across the sample volume, which could subsequently result in weaknesses in the polymer product. MRI has also been used to study porous materials such as ceramics and catalysts. In the latter example, the effects of manufacturing processes have been considered with respect to limiting heterogeneities in the pore structure, which may reduce the active lifetime of the catalyst.125 Pore structure has also been shown to be an important factor in the drying mechanism of porous alumina catalyst pellets.126 MRI has been utilised in the study of transport processes in packed columns and bioreactors.125,126 From such studies it may then be possible to optimise the method for packing chromatography columns129 and the design of membranes.130 Flow imaging has been used to determine flow rates52–54,131 and diffusion coefficients of liquids.123 Experiments have also been performed on two-phase systems123 such as solid–liquid suspensions, in an attempt to improve understanding of the rheology of these systems.
MRI has been applied extensively in the food industry to obtain both structural and dynamic information of a wide variety of foodstuffs.123,132 The internal structure of, for example, fruit and vegetables has been studied using this non-invasive technique.133 It has been shown in courgettes that, upon freezing, the cell walls rupture and the tissue morphology is altered.134 One-dimensional magnetic resonance projections have been obtained of pickled olives moving through the magnet on a conveyer belt.135 It was possible to distinguish between pitted and non-pitted olives. A similar study has also been performed on cherries.136 The quality of food has been studied in terms of its ripeness, maturity137 and bruising.138 MRI has also been used to show that, during the baking of a cookie,139 a gradient in moisture develops from the middle to the edge. The quality of cheese is assessed by examining its internal structure. This usually involves cutting the cheese in half so that the two cut surfaces can be examined. MRI offers a non-destructive method for examination of the cheese structure.140 A hydration study of barley using MRI has revealed that the outer grain layers initially take up the water, which is then redistributed internally.141
6 Instrumentation for process analysis by NMR spectrometry
Low-field NMR spectrometers have been used extensively in the food, polymer, petroleum and pharmaceuticals industries. Typical examples cited include density, viscosity and crystallinity measurements of polymers, rock core studies, and the measurement of the moisture content of powders, the fluoride content in toothpastes and the water and fat content in foods. The instruments employ a permanent magnet, with an operating frequency between 1 and 65 MHz for 1H (although 20 MHz is the typical operating frequency), and are designed for bench-top use. The instruments usually consist of a box containing the magnet and the electronics, and are operated using a personal computer. Instruments which operate at 1 or 2 MHz (designed for rock core studies) are in some cases small enough to fit into two suitcases, thus enhancing their portability. A number of instruments are commercially available including those manufactured by Resonance Instruments,78 Bruker (Minispec),79 Process Control Technology142 and Oxford Instruments.143 Although such instruments are frequently referred to as process analysers in the literature, their primary use is in quality control and process development.The information required from the analysis is derived from the FID (NMR signal in the time domain). Samples are analysed in standard NMR tubes although the diameter can range from the usual 5 mm to 60 mm to facilitate the analysis of viscous and/or relatively insensitive samples. All instruments are capable of carrying out 1H NMR experiments, and the majority can also analyse 19F. However, only those instruments with a relatively high magnetic field are capable of carrying out 31P NMR studies.
Process NMR analysers are currently manufactured by Foxboro144 (previously Elbit-ATI) and Oxford Instruments143 (previously Auburn). Both companies market instrumentation which is designed for use in a process environment, either in an analyser house or adjacent to the process stream. The Foxboro I/A Series NMR spectrometer has been used in the oil industry and has been in operation in a Texaco oil refinery in California since 1995. Foxboro have named many possible applications in oil refineries, including measurement of the aromatic and olefinic content of gasoline and diesel, the average chain length of components in crude oil, oxygenates concentration, density and to monitor the weight percent of acid in acid alkylation reactions. The instrument employs a permanent magnet (60 MHz for 1H), a LiCl lock and automatic shimming, and analysis is carried out in the ‘stopped-flow’ mode. The NMR signal obtained from the Foxboro analyser is Fourier transformed to yield spectral data, which are then analysed using partial least squares (PLS) calibration algorithms. An analyser manufactured by Oxford Instruments, MagModule 2000,143 has been used to determine the density and melt index of samples of polyethylene.
Intermagnetics manufacture a MRI spectrometer to permit non-contact and non-destructive testing of samples on a production line.145 The instrument employs a low-field permanent magnet. Such instruments have been used to detect leaks in plastic containers of soya milk and to determine ripeness of fruit. Recently, a ‘fist-sized’ NMR analyser146,147 has been marketed by Bruker79 to permit analysis of surfaces of arbitrarily shaped objects. This type of instrumentation has been used to obtain a depth profile of a rubber car tyre. A very recent, interesting development is the introduction of a high-field (300 MHz for 1H) NMR spectrometer, INCA, by Bruker.79 Although the instrument employs a superconducting magnet, the magnet is very well shielded. The entire instrument is contained within a single, transportable enclosure and operation of the spectrometer is highly automated. The instrument is designed for on-line process control and quality control analyses.
7 Chemometrics and data analysis
Traditionally, the FID (time domain) obtained in a NMR experiment is Fourier transformed to produce a spectrum (frequency domain). Quantitative information can then be obtained from the spectrum by integration of the NMR signals. This is the usual method for obtaining quantitative information from high-field NMR spectra. For example, agricultural chemicals have been analysed quantitatively by integration of high-field 1H and 31P NMR spectra, with an accuracy and precision of the order of 0.5%.148 However, even for those spectra acquired on high-field instruments, some overlap of resonances is likely. Hence, commercially available NMR spectrometer software usually contains some sort of deconvolution program. At low magnetic fields, due to the decrease in spectral resolution, the problem of overlapping resonances is accentuated. This can result in spectra that are quite complicated and difficult to interpret visually, particularly in the case of 1H NMR spectra. In such cases, chemometric techniques may be able to help in data interpretation. Chemometric techniques149–155 can be used to extract the desired information from multivariate data. Chemometrics comprises the application of multivariate statistics, mathematics and computational methods to chemical measurements. The central idea is that mathematical and statistical methods can be used to enhance the productivity of chemical experimentation. In this section, methods which have been used to analyse high-field NMR data (in the time or frequency domain), that could have potential use in the analysis of low-field process NMR data, are discussed. These include procedures for in vivo analysis of biological systems, solid-state studies and imaging.Multivariable analysis has been applied to 13C NMR spectra of olive oils in an attempt to differentiate between olive oils from different regions based on their spectra.156 Principal component analysis (PCA) scores could be used to discriminate between different varieties of olive oil. Using partial least squares (PLS), it was possible to predict the variety of an olive oil for over 90% of the test oils.
Pattern recognition methods based on artificial neural networks have been used to analyse 1H NMR spectra of binary alditol mixtures.157 From analysis of the 3.5 to 4.0 ppm spectral region, it was possible to classify spectra of mixtures and also to obtain quantitative estimates of the relative concentrations of the two components in the mixtures. Spectra with a low signal-to-noise ratio and of poorer resolution were also analysed, as the artificial neural networks were trained to tolerate variations in the input quality of the data. It was also possible to identify components present at concentrations greater than 10% in more complex mixtures (greater than two components) of the alditols.
Chemometric techniques have been used in the simulation of NMR spectra. For example, 13C NMR spectra were simulated for methyl-substituted norborn-2-ol compounds.149 In the simulations, terms were included for bond distances, torsional angles and van der Waals interactions. From the so-called reference set of compounds, spectra were then successfully predicted for related compounds. Similar studies have also been performed on dibenzofurans,158 where multiple linear regression analysis and neural networks were used to simulate spectra of toxic compounds. This enabled the structure of such compounds to be predicted, thus negating the need for handling them. Principal component analysis (PCA) and partial least squares (PLS) were applied to 13C CP/MAS (cross polarisation with magic-angle spinning) spectra of lignocellulosic to estimate the amount of different cellulose polymorphs present.159
Samples of crude oil have been analysed using 13C NMR spectrometry.160 Using PCA, it was possible to correlate samples obtained from the same geographical area. PCA has also been used to determine the composition of styrene/butadiene copolymers from 13C NMR spectra.161 The results obtained using PCA were comparable to those from direct analysis of 13C NMR spectra.
Abstract factor analysis has been used to extract component information in solid-state NMR spectra that exhibit overlapping powder patterns, or in the case of spectra acquired with magic-angle spinning (MAS), overlapping sidebands.162 Phosphorus-31 spectra were obtained of octacalcium phosphate using different cross polarisation contact times, in order to yield factor-analysable chemical shift mixtures.
Gel-permeation chromatography (GPC)-NMR spectrometry allows correlation of molecular weight distribution with chemical information. However, GPC analysis of polymer blends gives chromatograms of poor resolution. Similarly, broad 1H NMR spectra are obtained of polymer blends. Multivariate curve resolution has been used to resolve GPC-NMR spectra of an acrylic acid and laurylmethylacrylic acid copolymer.163 Using this technique, it was possible to resolve two different components, which can be attributed to the extended and collapsed forms of the polymer.
The wavelet transform method can be used to quantify the chemical shift, transverse relaxation time (T2), amplitude and phase of a NMR signal in the time domain.164,165 With one of the proposed methods,165 noisy, overlapping 31P NMR signals obtained from perfused muscle were successfully analysed. Blood plasma is particularly difficult to analyse using 1H NMR spectrometry as the spectra contain numerous overlapping signals. However, through the use of wavelet transform time domain processing, it has been possible to analyse quantitatively the methylene spectral region of blood plasma lipoprotein fractions.166,167 The results obtained using line shape fitting of the frequency domain and wavelet transform analysis of the FID were compared for 1H NMR data obtained from, plasma lipoproteins.168 In this study, it was suggested that the wavelet transform method was preferable to line-shape fitting, as assumptions concerning the number of signals needed for an adequate description of the data were not needed. The wavelet transform has been used to analyse quantitatively a broad underlying resonance, as well as the other narrower resonances, in the 31P NMR spectrum of gerbil brain.167 It has also been used to resolve solid-state 13C NMR spectra of organic molecules from those of the resin in solid-state support organic synthesis.167 It has also been used to remove large unwanted signals from spectra.47,164,169
A singular value decomposition algorithm has been used to determine the number of exponentials present in a FID.170 Transverse relaxation times can then be calculated for, for example, sunflower oils and water samples containing different concentrations of MnCl2.
Factor analysis has also been carried out on 19F time domain data, generated using a solid-echo, of poly(tetrafluoroethene) (PTFE).171 It was possible to correlate the second factor with crystallinity, which was determined using XRD.
Multivariate regression has been used to determine the moisture content in meat products from the FID.172
Data have been simulated to represent the type of signals that might be expected from a process NMR analyser.173 FIDs were simulated, using data obtained for polymer samples, for three component mixtures. In one data set, only the amplitude values were varied, and T2 values were fixed, whereas in a second data set both sets of parameters were varied. PLS calibration models were built to predict the amplitudes and also the relaxation time parameters of individual components. The effects of signal-to-noise ratio and errors in reference measurements on the predictive accuracy of the PLS algorithm were examined, and the latter found to have the greater effect.
Margarine and butter samples have been analysed using low-field 1H NMR spectrometry.174 Surface plots were constructed from T1 and T2 values, which were then analysed using ANOVA (analysis of variance) to determine whether the data contained information relating to moisture content. PLS calibration models were then built from those data sets that contained such information. Factor analysis and ANOVA have also been used to analyse 1H NMR data of spreads and gelatines.175
A number of methods that involve analysis of the FID have found favour in the analysis of in vivo31P NMR data, which often includes signals that are overlapped and of poor signal-to-noise ratio. The methods proposed can be divided into two categories; non-interactive and interactive. Non-interactive methods start by assuming that the FID is composed of a sum of exponentially damped sinusoids. For the non-interactive procedures, linear prediction (LP) or state-space methods (involving formation of a Hankel matrix from the data) in combination with singular value decomposition (SVD) have been employed. The parameters of interest, i.e., frequencies, amplitudes, transverse relaxation times and phases of signals, can then be determined using a least squares (LS) or total least squares (TLS) procedure. Spectral reconstruction from the calculated parameters can give rise to a de-noised spectrum, as at the SVD stage there is an opportunity for separation of signal from noise. Using linear prediction, SVD and a least squares procedure (termed LPSVD),176 it was possible to determine the four parameters for all eight signals in a 31P FID of an anaesthetised mouse using data points 4 to 128 of the 768 points acquired. A method involving formation of a Hankel matrix, SVD and a total least squares procedure (HTLS), has been used to analyse the 31P FID of a perfused rat liver.177,178 Accurate quantification of the orthophosphate region of the spectra acquired from liver, brain and muscle is difficult due to the large number of overlapping signals (inorganic phosphate, phosphomonoesters and phosphodiesters).
The non-interactive methods described above do not allow for the incorporation of any knowledge into the signal analysis stage. It has been shown that, although the overall fit of these methods can be good even for overlapping signals with a poor signal-to-noise ratio, the fitted parameters do not accurately fit individual components. Incorporation of information into the model, such as that relating to the number of spectral lines and frequencies, can dramatically improve the precision of the results. A variable projection (VARPRO) algorithm has been used for this purpose.179 VARPRO has also been used to analyse quantitatively the C(1) signal in the 13C NMR spectrum (acquired with 1H decoupling) of glycogen.180 The signal is not a single Lorentzian, but is in fact a superposition of a number of lines centred at the same frequency, but with different line widths. The results obtained using VARPRO were found to be superior to integration methods, an iterative line-fitting procedure in the frequency domain and to those obtained using HTLS.
HTLS has been adapted to allow prior knowledge (HTLS-PK) concerning known signal poles to be incorporated into the model.181 The 31P FID of a brain of a healthy volunteer was analysed using this method. As the spectrum contains a number of overlapping peaks, one of which is particularly small, it was possible to improve quantification through the use of HTLS-PK.
Continuous regularisation (CR) can be used to determine the number of spectral components.182 Hence, in combination with LPSVD [LPSVD(CR)], it is possible to automate the determination of the number of signals, and subsequently determine their parameters. For example, using LPSVD(CR), it was possible to determine the correct number of components in the 31P signals of a human calf muscle and brain. Interpretation of such NMR signals is challenging as the former includes low intensity signals, whereas the latter contains a narrow inorganic phosphate peak in between a broad phosphomonoester and a very broad phosphodiester peak.
Another interactive method, which was found to be superior to VARPRO, is AMARES (advanced method for accurate, robust and efficient spectral fitting).183 The AMARES method has also been used to analyse simultaneously a series of 31P FIDs from a kinetics experiment.184 Prior knowledge in the form of frequency information can be included in, for example, the HTLS procedure by extracting the signals of interest from the overall FID using a highly selective finite impulse response (FIR) filter.185,186 This method has also been combined with a variant of AMARES, an iterative non-linear least squares (NLLS) algorithm.187 The RELAX algorithm has been adapted to include information relating to possible frequency intervals of the signals.188
Pulsed gradient spin-echo (PGSE) NMR experiments have been used to resolve spectra of individual components in multicomponent solutions.189 Through the use of a procedure called the direct exponential curve resolution algorithm (DECRA), it was possible to isolate the 1H NMR spectrum (at 400 MHz) of gelatin from that of a non-ionic surfactant. In a similar way, relaxation times have been used in multivariate image analysis.190–192 It was possible to separate the two components that made up a phantom using the fact that one component NiCl2(aq) had a spin–spin relaxation time (T2) that was five times that of the other component [MnCl2(aq)].190 DECRA has also been used to resolve MR images of the human brain.191–193 Mixtures of polymorphs have been analysed using solid-state 13C NMR and DECRA.194,195 DECRA was able to extract individual spectra from a mixture spectrum, due to the fact that the two different polymorphs had different 1H T1 values.
A combination of 13C NMR spectrometry and multivariate data analysis has been used to identify individual constituents in model hydrocarbon mixtures and also in real petroleum fractions.196 It was found that a single principal component described 85–90% of the chemical shift variance. From analysis of the loading plot, it was possible to deduce that this could be accounted for by variations in the aromaticity of the molecule. Hence, it was necessary to reference separately aromatic methine, saturated methine/methylene and methyl carbons.
Both 1H and 13C NMR spectrometries (at 400 MHz) have been used to classify wines according to their origin.197 However, it was necessary to pre-process the data using a technique called partial linear fit198 to remove variations in the observed chemical shifts due to solvent, temperature and magnetic field homogeneity effects. It was concluded that, where possible, 1H NMR spectrometry should be used to classify wines, as acquisition of 13C NMR spectra takes considerably longer. The most common method for correction of chemical shift variations due to, for example, solvent effects, is to simply degrade the resolution of the spectrum. This method is satisfactory for small variations in chemical shift, but is poor for complex spectra and cases where shifts are large compared to the separation of signals.198
8 Conclusions
NMR spectrometry has many advantages in process applications including the possibility of standardless quantitative analysis. Most measurements at low fields have been based on the analysis of the free induction decay signal to give information on physical parameters or the moisture/fat content of materials. However, reports of chemical composition analysis by low-field 1H NMR spectrometry are increasing. Flow NMR spectrometry could find several useful applications in process analysis, and procedures devised for LC-NMR studies may be useful for the on-line monitoring of manufacturing processes. Recent developments in signal processing and chemometric procedures are likely to provide significant benefits in the interpretation of the convoluted spectra often produced by low-field NMR spectrometry. In particular, mathematical procedures devised to aid the analysis of NMR spectra in medical applications may prove equally as useful in the chemical monitoring of process streams and liquors. Although 1H NMR signals are more sensitive, 19F and 31P NMR measurements may be beneficial in studies of F- or P-containing compounds, especially when hydrocarbon solvents are used. Process NMR spectrometry will not be suitable for trace analysis, but in many applications it may be a useful alternative to established procedures for quantitative monitoring of batch and continuous reactions.9 Acknowledgements
AN’s fellowship was funded as part of the Foresight Challenge award from the Office of Science and Technology to establish CPACT. CMcG acknowledges CPACT for the award of a studentship.References
- J. Workman, Jr., D. J. Veltkamp, S. Doherty, B. B. Anderson, K. E. Creasy, M. Kock, J. F. Tatera, A. L. Robinson, L. Bond, L. W. Burgess, G. N. Bokerman, A. H. Ullman, G. P. Darsey, F. Mozayeni, J. A. Bamberger and M. S. Greenwood, Anal. Chem., 1999, 71, 121R CrossRef.
- K. R. Beebe, W. W. Blaser, R. A. Bredeweg, J. P. Chauvel, Jr., R. S. Harner, M. A. LaPack, A. Leugers, D. P. Martin, L. G. Wright and E. D. Yalvac, Anal. Chem., 1993, 65, 199R CAS.
- W. W. Blaser, R. A. Bredeweg, R. S. Harner, M. A. LaPack, A. Leugers, D. P. Martin, R. J. Bell, J. Workman, Jr. and L. G. Wright, Anal. Chem., 1995, 67, 47R CAS.
- C. Tellier and F. Mariette, Ann. Rep. NMR Spectrosc., 1995, 31, 105 Search PubMed.
- J. F. Haw and T. W. Skloss, Spectroscopy, 1993, 8, 22 Search PubMed.
- T. W. Skloss, A. J. Kim and J. F. Haw, Anal. Chem., 1994, 66, 536 CrossRef CAS.
- R. M. Pearson, L. R. Ream and J. Adams, Cereal Foods World, 1987, 32, 822 Search PubMed.
- D. Neuhaus, Nuclear Overhauser Effect, Encyclopedia of NMR, ed. D. M. Grant and R. K. Harris, Wiley, London, 1996, 5, 3290. Search PubMed.
- R. R. Ernst, G. Bodenhausen and A. Wokaun, Principles of Nuclear Magnetic Resonance in One and Two Dimensions, Oxford University Press, 1991. Search PubMed.
- D. A. Laude, Jr. and C. L. Wilkins, Macromolecules, 1986, 19, 2295 CrossRef.
- G. Suryan, Proc. Indian Acad. Sci. A, 1951, 33, 107 Search PubMed.
- E. Bayer and K. Albert, J. Chromatogr., 1984, 312, 91 CrossRef CAS.
- J. F. Haw, T. E. Glass, D. W. Hausler, E. Motell and H. C. Dorn, Anal. Chem., 1980, 52, 1135 CrossRef CAS.
- L. R. Hirschel and L. F. Libelo, J. Appl. Phys., 1961, 32, 1404.
- A. I. Zhernovi and G. D. Latyshev, Nuclear Magnetic Resonance in a Flowing Liquid, Consultants Bureau, New York, 1965. Search PubMed.
- D. A. Laude, Jr., R. W. K. Lee and C. L. Wilkins, Anal. Chem., 1985, 57, 1287.
- J. F. Haw, T. E. Glass and H. C. Dorn, J. Magn. Reson., 1982, 49, 22 CAS.
- M. Godejohann, A. Preiss and C. Mügge, Anal. Chem., 1998, 70, 590 CrossRef CAS.
- D. A. Laude, Jr., R. W. K. Lee and C. L. Wilkins, Anal. Chem., 1985, 57, 1281 CrossRef.
- D. A. Laude, Jr., R. W. K. Lee and C. L. Wilkins, J. Magn. Reson., 1984, 60, 453.
- M. C. McIvor, J. Phys. E, 1969, 2, 292 Search PubMed.
- S. A. Korhammer and A. Bernreuther, Fresenius’ J. Anal. Chem., 1996, 354, 131 CAS.
- L. Griffiths, Anal. Chem., 1995, 67, 4091 CrossRef CAS.
- S. Stevenson, T. Glass and H. C. Dorn, Anal. Chem., 1998, 70, 2623 CrossRef CAS.
- U. L. Günther, J. L. Sudmeier, K. Albert and W. W. Bachvchin, J. Magn. Reson. A, 1995, 117, 73 CrossRef.
- D. Wu and C.S. Johnson, Jr., J. Magn. Reson., 1997, 127, 225 CrossRef CAS.
- D. W. Jones and T. F. Child, Adv. Magn. Reson., 1976, 31, 123 Search PubMed.
- J. L. Sudmeier, U. L. Günther, K. Albert and W. W. Bachovchin, J. Magn. Reson. A, 1996, 118, 145 CrossRef CAS.
- H. Barjat, G. A. Morris, M. J. Newman and A. G. Swanson, J. Magn. Reson. A, 1996, 119, 115 CrossRef CAS.
- K. Albert and E. Bayer, HPLC detection: Newer methods, ed. G. Patonay, VCH, New York, ch. 9, p. 197. Search PubMed.
- J. F. Haw, T. E. Glass and H. C. Dorn, Anal. Chem., 1981, 53, 2327 CrossRef CAS.
- J. F. Haw, T. E. Glass and H. C. Dorn, Anal. Chem., 1981, 53, 2332 CrossRef CAS.
- J. Buddrus and H. Herzog, Org. Magn. Reson., 1980, 13, 153 Search PubMed.
- J. C. Lindon, J. K. Nicholson and I. D. Wilson, Prog. NMR Spectrosc., 1996, 29, 1 Search PubMed.
- J. C. Lindon, J. K. Nicholson and I. D. Wilson, J. Chromatogr. B, 2000, 748, 233 CrossRef CAS.
- M. Spraul, M. Hofmann, P. Dvortsak, J. K. Nicholson and I. D. Wilson, J. Pharm. Biomed. Anal., 1992, 10, 601 CrossRef CAS.
- H. Pasch and W. Hiller, Macromolecules, 1996, 29, 6556 CrossRef CAS.
- H. Pasch, W. Hiller and R. Haner, Polymer, 1998, 39, 1515 CrossRef CAS.
- M. Godejohann, A. Preiss, C. Mügge and G. Wünsch, Anal. Chem., 1997, 69, 3837 CrossRef CAS.
- N. Watanabe and E. Niki, Proc. Japan Acad. B, 1978, 54, 194 Search PubMed.
- E. Bayer, K. Albert, M. Nieder, E. Grom and T. Keller, J. Chromatogr. A, 1979, 186, 497 CrossRef CAS.
- B. Behnke, G. Schlotterbeck, U. Tallarek, S. Strohschein, L. H. Tseng, T. Keller, K. Albert and E. Bayer, Anal. Chem., 1996, 68, 1110 CrossRef CAS.
- D. A. Laude, Jr. and C. L. Wilkins, Anal. Chem., 1987, 59, 546 CrossRef.
- D. A. Laude, Jr., R. W. K. Lee and C. L. Wilkins, Anal. Chem., 1985, 57, 1464 CrossRef CAS.
- W. S. Price, Ann. Rep. NMR Spectrosc., 1999, 38, 289 Search PubMed.
- T. Sundin, L. Vanhamme, P. Van Hecke, I. Dologou and S. Van Huffel, J. Magn. Reson., 1999, 139, 189 CrossRef CAS.
- H. Serrai, L. Senhadji, D. Clayden and J. D. de Certaines, Spectrosc. Lett., 2000, 33, 47 CAS.
- K. Albert, U. Braumann, L. H. Tseng, G. Nicholson, E. Bayer, M. Spraul, M. Hofmann, C. Dowle and A. M. Chippendale, Anal. Chem., 1994, 66, 3042 CrossRef CAS.
- U. Braumann, H. Händel, K. Albert, R. Ecker and M. Spraul, Anal. Chem., 1995, 67, 930 CrossRef CAS.
- K. Albert, J. Chromatogr. A, 1997, 785, 65 CrossRef CAS.
- L. A. Allen, M. P. Spratt, T. E. Glass and H. C. Dorn, Anal. Chem., 1988, 60, 675 CrossRef CAS.
- A. Caprihan and E. Fukushima, Phys. Rep., 1990, 198, 195 CrossRef CAS.
- T. Q. Li, J. D. Seymour, R. L. Powell, K. L. McCarthy, L. Ödberg and M. J. McCarthy, Magn. Reson. Imaging, 1994, 12, 923 CrossRef CAS.
- K. D. Merboldt, W. Hänicke and J. Frahm, J. Magn. Reson., 1986, 67, 336.
- C. A. Fyfe, M. Cocivera and S. W. H. Damji, Acc. Chem. Res., 1978, 11, 271 CrossRef.
- C. A. Fyfe, M. Cocivera, S. W. H. Damji, T. A. Hostetter, D. Sproat and J. O’Brien, J. Magn. Reson., 1976, 23, 377 CAS.
- Y. Asahi and E. Mizuta, Talanta, 1972, 19, 567 CrossRef CAS.
- S. Funahashi, K. Ishihawa, S. Aizawa, T. Sugata, M. Ishii, Y. Inada and M. Tanaka, Rev. Sci. Instrum., 1993, 64, 130 CrossRef CAS.
- J. A. Iggo, D. Shirley and N. C. Tong, New J. Chem., 1998, 1043 RSC.
- M. Spraul, M. Hofmann, M. Ackermann, A. W. Nicholls, S. J. P. Damment, J. N. Haselden, J. P. Shockcor, J. K. Nicholson and J. C. Lindon, Anal. Commun., 1997, 34, 339 RSC.
- P. J. Hore, S. L. Winder, C. H. Roberts and C. M. Dobson, J. Am. Chem. Soc., 1997, 119, 5049 CrossRef CAS.
- M. Hunger and T. Horvath, Chem. Commun., 1995, 1423 RSC.
- P. Goguen and J. F. Haw, J. Catalysis, 1996, 161, 873 Search PubMed.
- M. E. Lacey, R. Subramanian, D. L. Olson, A. G. Webb and J. V. Sweedler, Chem. Rev., 1999, 99, 3133 CrossRef CAS.
- N. Wu, T. L. Peck, A. G. Webb, R. L. Magin and J. V. Sweedler, Anal. Chem., 1994, 22, 3849 CrossRef.
- D. L. Olson, M. E. Lacey, A. G. Webb and J. V. Sweedler, Anal. Chem., 1999, 71, 3070 CrossRef CAS.
- R. Subramanian, W. P. Kelley, P. D. Floyd, Z. J. Tan, A. G. Webb and J. V. Sweedler, Anal. Chem., 1999, 71, 5335 CrossRef CAS.
- E. MacNamara, T. Hou, G. Fisher, S. Williams and D. Raftery, Anal. Chim. Acta, 1999, 397, 9 CrossRef CAS.
- T. Hou, E. MacNamara and D. Raftery, Anal. Chim. Acta, 1999, 400, 297 CrossRef CAS.
- G. Fisher, C. Petucci, E. MacNamara and D. Raftery, J. Magn. Reson., 1999, 138, 160 CrossRef CAS.
- Y. Li, A. M. Wolters, P. V. Malawey, J. V. Sweedler and A. G. Webb, Anal. Chem., 1999, 71, 4815 CrossRef CAS.
- R. L. Haner, W. Llanos and L. Mueller, J. Magn. Reson., 2000, 143, 69 CrossRef CAS.
- G. E. Maciel, NMR in industrial process control and quality control, Nuclear Magnetic Resonance in Modern Technology, ed. G. E. Maciel, Kluwer Academic, Netherlands, 1994, p. 225. Search PubMed.
- T. M. Shaw and R. H. Elsken, J. Chem. Phys., 1953, 21, 565 CrossRef CAS.
- W. Derbyshire, Applications of low-resolution NMR, Analytical Applications of Spectroscopy, ed. C. S. Creaser and A. M. C. Davies, Royal Society of Chemistry, London, 1988, p. 325. Search PubMed.
- R. B. Wettstrom, J. Am. Oil Chem. Soc., 1971, 48, 15 Search PubMed.
- T. F. Conway, J. Am. Oil Chem. Soc., 1971, 48, 54 Search PubMed.
- Resonance Instruments, www.resonance.co.uk.
- Bruker NMR, www.bruker.com.
- C. Job, R. M. Pearson and M. F. Brown, Rev. Sci. Instrum., 1994, 65, 3354 CrossRef CAS.
- L. S. Simeral and P. H. Krygsman, Appl. Spectrsoc., 1999, 53, 1009 Search PubMed.
- E. W. Tollner and W. L. Rollwitz, Trans. ASAE, 1988, 31, 1608 Search PubMed.
- R. F. Paetzold, G. A. Matzkanin and A. De Los Santos, Soil Sci. Soc. Am. J., 1985, 49, 537 Search PubMed.
- R. F. Paetzold, A. De Los Santos and G.A. Matzkanin, Soil Sci. Soc. Am. J., 1987, 51, 287 Search PubMed.
- M. C. Vackier and D. N. Rutledge, J. Magn. Reson. Anal., 1996, 2, 311 Search PubMed.
- M. C. Vackier, A. S. Barros and D. N. Rutledge, J. Magn. Reson. Anal., 1996, 2, 321 Search PubMed.
- M. C. Vackier, B. P. Hills and D. N. Rutledge, J. Magn. Reson., 1999, 138, 36 CrossRef CAS.
- I. Fourel, J. P. Guillement and D. Le Botlan, J. Colloid Interface Sci., 1994, 164, 48 CrossRef CAS.
- I. Fourel, J. P. Guillement and D. Le Botlan, J. Colloid Interface Sci., 1995, 169, 119 CrossRef CAS.
- S. I. Cho and C. H. Chung, Trans. ASAE, 1997, 40, 1129 Search PubMed.
- J. P. Monteiro Marques, D. N. Rutledge and C. J. Ducauze, Int. J. Food Sci. Technol., 1991, 26, 173.
- E. Brosio, G. Altobelli and A. Di Nola, J. Food Technol., 1984, 19, 103 Search PubMed.
- E. Brosio, G. Altobelli, Y. Shi Yun and A. Di Nola, J. Food Technol., 1983, 18, 219 Search PubMed.
- E. Brosio, F. Conti, A. Di Nola, M. Scalzo and E. Zulli, J. Am. Oil Chem. Soc., 1982, 59, 59 Search PubMed.
- M. Guillou and C. Tellier, Anal. Chem., 1988, 60, 2182 CrossRef CAS.
- G. E. Renzoni, E. G. Shankland, J. A. Gaines and J. B. Callis, Anal. Chem., 1985, 57, 2864 CrossRef CAS.
- C. L. Glaves, P. J. Davies and D. M. Smith, Powder Technol., 1988, 54, 261 CrossRef CAS.
- P. Desbois and D. Le Botlan, J. Food Sci., 1994, 59, 1088 Search PubMed.
- G. Roudaut, D. van Dusschoten, H. Van As, M. A. Hemminga and M. Le Meste, J. Cereal Sci., 1998, 28, 147 CrossRef CAS.
- F. Capozzi, M. A. Cremonini, C. Luchinat, G. Placucci and C. Vignali, J. Magn. Reson., 1999, 138, 277 CrossRef CAS.
- F. Mariette and P. Marchal, J. Magn. Reson. Anal., 1996, 2, 290 Search PubMed.
- D. A. Netzel and T. F. Turner, Fuel Sci. Technol. Int., 1990, 8, 379 Search PubMed.
- A. L. Newbury, R. B. Lowry and D. Short, Adv. Composites Lett., 1995, 4, 189 Search PubMed.
- P. Lambelet, R. Berrocal, C. Desarzens, I. Froehlicher and F. Ducret, J. Food Sci., 1988, 53, 943 Search PubMed.
- F. Schierbaum, S. Radosta, W. Vorweg, Y. P. Yuriev, E. E. Braudo and M. L. German, Carbohydrate Polymers, 1992, 118, 155 Search PubMed.
- W. R. Ladner and A. E. Stacey, Br. J. Appl. Phys., 1962, 13, 136 Search PubMed.
- J. D. King and W. L. Rollwitz, ISA Trans., 1983, 22, 69 CAS.
- C. I. Nicholls and A. De Los Santos, Drying Technol., 1991, 9, 849 Search PubMed.
- C. Tellier, M. Trierweiler, J. Lejot and G. J. Martin, Analusis, 1990, 18, 167 Search PubMed.
- R. M. Pearson and D. L. Wetzel, Cereal Foods World, 1985, 30, 563 Search PubMed.
- R. M. Pearson, Cereal Foods World, 1987, 32, 653 Search PubMed.
- R. M. Pearson and L. R. Ream, Cereal Foods World, 1987, 32, 658 Search PubMed.
- R. M. Pearson, L. Ryhtia and C. Job, J. Metals, 1985, 37, A23 Search PubMed.
- R. M. Pearson and T. L. Parker, J. Metals, 1983, 35, A4 Search PubMed.
- R. M. Pearson, J. Metals, 1982, 35, A34 Search PubMed.
- S. B. Thoma, D. M. Smith, J. Boughton and R. Davies, Part. Part. Syst. Charact., 1993, 10, 246 CrossRef CAS.
- M. L. Snoddy, Spectroscopy, 1983, 8, 41 Search PubMed.
- F. A. Nelson, C. A. Reilly and W. E. Savage, Indust. Eng. Chem., 1960, 52, 487 Search PubMed.
- T. W. Skloss, Ph.D. Thesis, Texas A&M University, USA, 1996..
- J. C. Edwards and P. J. Giammatteo, Proc. Annual ISA Anal. Div. Symp., 1998, 31, 73 Search PubMed.
- R. Neudert, E. Ströfer and W. Bremser, Magn. Reson. Chem., 1986, 24, 1089 CAS.
- E. J. Draheim and A. J. Ragauskas, J. Wood Chem. Technol., 1997, 17, 287 Search PubMed.
- L. F. Gladden, Chem. Eng. Sci., 1994, 49, 3339 CrossRef CAS.
- L. D. Hall and T. A. Carpenter, Magn. Reson. Imaging, 1992, 10, 713 CrossRef CAS.
- L. F. Gladden and P. Alexander, Meas. Sci. Technol., 1996, 7, 423 CrossRef CAS.
- L. F. Gladden, Chem. Eng. J. Biochem. Eng. J., 1995, 56, 149 CrossRef CAS.
- P. Jezzard, C. J. Wiggins, T. A. Carpenter, L. D. Hall, P. Jackson, N. J. Clayden and N. J. Walton, Adv. Materials, 1992, 4, 82 Search PubMed.
- P. Jackson, N. J. Clayden, N. J. Walton, T. A. Carpenter, L. D. Hall and P. Jezzard, Polymer Int., 1991, 24, 139 Search PubMed.
- E. Bayer, W. Müller, M. Ilg and K. Albert, Angew. Chem. Int. Ed. Engl., 1989, 28, 1029 CrossRef.
- C. A. Heath, G. Belfort, B. E. Hammer, S. D. Mirer and J. M. Pimbley, A.I.Ch.E. J., 1990, 36, 547 Search PubMed.
- D. F. Arola, G. A. Barrall, R. L. Powell, K. L. McCarthy and M. J. McCarthy, Chem. Eng. Sci., 1997, 52, 2049 CrossRef CAS.
- M. J. McCarthy and K. L. McCarthy, Magn. Reson. Imaging, 1996, 14, 799 CrossRef CAS.
- P. Chen, M. J. McCarthy and R. Kauten, Trans. ASAE, 1989, 32, 1747 Search PubMed.
- S. L. Duce, T. A. Carpenter and L. D. Hall, J. Food Eng., 1992, 16, 165 CrossRef.
- B. Zion, S. M. Kim, M. J. McCarthy and P. Chen, J. Sci. Food Agric., 1997, 75, 496 CrossRef CAS.
- B. Zion, M. J. McCarthy and P. Chen, Food Sci. Technol., 1994, 27, 457 Search PubMed.
- P. Chen, M. J. McCarthy, R. Kauten, Y. Sarig and S. Han, J. Agric. Eng. Res., 1993, 55, 177 CrossRef.
- B. Zion, P. T. Chen and M. J. McCarthy, Comput. Electron. Agric., 1995, 13, 289 CrossRef.
- S. W. Hong, Z. Y. Yan, M. S. Otterburn and M. J. McCarthy, Magn. Reson. Imaging, 1996, 14, 923 CrossRef CAS.
- S. M. Kim, M. J. McCarthy and P. Chen, J. Magn. Reson. Anal., 1996, 2, 281 Search PubMed.
- E. McEntyre, R. Ruan and R. G. Fulcher, Cereal Chem., 1998, 75, 792 Search PubMed.
- Process Technology Control Corporation, www.pctnmr.com.
- Oxford Instruments, www.oxford-america.com/analytical.
- Foxboro, www.foxboro.com.
- Intermagnetics General Coroporation, www.igc.com.
- G. Eidmann, R. Savelberg, P. Blümler and B. Blümich, J. Magn. Reson. A, 1996, 122, 104 CrossRef CAS.
- A. Guthausen, G. Zimmer, P. Blümler and B. Blümich, J. Magn. Reson., 1998, 130, 1 CrossRef CAS.
- G. Maniara, K. Rajamoorthi, S. Rajan and G. W. Stockton, Anal. Chem., 1998, 70, 4921 CrossRef CAS.
- P. C. Jurs, Rev. Comput. Chem., 1990, 169 Search PubMed.
- I. E. Frank and B. R. Kowalski, Anal. Chem., 1982, 54, 232R CAS.
- L. S. Ramos, K. R. Beebe, W. P. Carey, E. Sánchez M., B. C. Erickson, B. E. Wilson, L. E. Wangen and B. R. Kowalski, Anal. Chem., 1986, 58, 294R CAS.
- S. D. Brown, S. T. Sum, F. Despagne and B. K. Lavine, Anal. Chem., 1996, 68, 21R CrossRef.
- Handbook of Chemometrics and Qualimetrics, Part A, ed. D. L. Massart, B. G. M. Vandeginste, L. M. C. Buydens, S. De Jong, P. J. Lewi and J. Smeyers-Verbeke, Elsevier, Amsterdam, 1997. Search PubMed.
- Handbook of Chemometrics and Qualimetrics, Part B, ed. D. L. Massart, B. G. M. Vandeginste, L. M. C. Buydens, S. De Jong, P. J. Lewi and J. Smeyers-Verbeke, Elsevier, Amsterdam, 1998. Search PubMed.
- R. G. Brereton, Analyst, 1987, 112, 1635 RSC.
- A. D. Shaw, A. di Camillo, G. Vlahov, A. Jones, G. Bianchi, J. Rowland and D. B. Kell, Anal. Chim. Acta, 1997, 348, 357 CrossRef CAS.
- S. R. Amendolia, A. Doppiu, M. L. Ganadu and G. Lubinu, Anal. Chem., 1998, 70, 1249 CrossRef CAS.
- D. L. Clouser and P. C. Jurs, Anal. Chim. Acta, 1996, 321, 127 CrossRef CAS.
- H. Lennholm, Abs. Pap. Amer. Chem. Soc., 1996, 212, 107-CELL Search PubMed.
- O. M. Kvalheim, D. W. Aksnes, T. Brekke, M. O. Eide, E. Sletten and N. Telnaes, Anal. Chem., 1985, 57, 2858 CrossRef CAS.
- N. G. Walsh, J. K. Hardy and P. L. Rinaldi, Appl. Spectrsoc., 1997, 51, 889 Search PubMed.
- D. W. Kormos and J. S. Waugh, Anal. Chem., 1983, 55, 633 CrossRef CAS.
- L. C. M. Van Gorkom and T. M. Hancewicz, J. Magn. Reson., 1998, 130, 125 CrossRef CAS.
- D. Barache, J.-P. Antoine and J. M. Dereppe, J. Magn. Reson., 1997, 128, 1 CrossRef CAS.
- H. Serrai, L. Senhadji, J. D. de Certaines and J. L. Coatrieux, J. Magn. Reson., 1997, 124, 20 CrossRef CAS.
- H. Serrai, L. Nadal, G. Leray, B. Leroy, B. Delplanque and J. D. de Certaines, NMR Biomed., 1998, 11, 273 CrossRef CAS.
- H. Serrai, L. Nadal, M. Le Floch, G. Leray, L. Senhadji, N. Le Tallec and J. D. de Certaines, J. Magn. Reson. Anal., 1997, 3, 79 Search PubMed.
- J. D. de Certaines, L. Nadal, G. Leray, H. Serrai and C. J. Lewa, Anticancer Res., 1996, 16, 1451 Search PubMed.
- N. Delprat, B. Escudié, P. Guillemain, R. Kronland-Martinet, P. Tchamitchian and B. Torrésani, IEEE Trans. Inform. Theory, 1992, 38, 644 CrossRef.
- M. Lupu and D. Todor, Chemom. Intell. Lab. Syst., 1995, 29, 11 CrossRef CAS.
- N. J. Clayden, Anal. Chim. Acta, 1997, 344, 261 CrossRef CAS.
- A. Gerbanowski, D. N. Rutledge, M. H. Feinberg and C. J. Ducauze, Sci. Aliments, 1997, 17, 309 Search PubMed.
- X. Lai, M. Sardashti, B. J. Lane, J. J. Gislason and D. J. O’Donnell, Appl. Spectrosc., 2000, 54, 54 Search PubMed.
- D. N. Rutledge and A. S. Barros, Analyst, 1998, 123, 551 RSC.
- D. N. Rutledge, A. S. Barros and F. Gaudard, Magn. Reson. Chem., 1997, 35, S13 CrossRef CAS.
- H. Barkhuijsen, R. de Beer, W. M. M. J. Bovée and D. van Ormondt, J. Magn. Reson., 1985, 61, 465 CAS.
- H. Chen, S. Van Huffel, C. Decanniere and P. Van Hecke, J. Magn. Reson. A, 1994, 109, 46 CrossRef CAS.
- S. Van Huffel, H. Chen, C. Decanniere and P. Van Hecke, J. Magn. Reson. A, 1994, 110, 228 CrossRef CAS.
- C. Decanniere, P. Van Hecke, F. Vanstapel, H. Chen, S. Van Huffel, C. van der Voort, B. van Tongeren and D. van Ormondt, J. Magn. Reson. B, 1994, 105, 31 CrossRef CAS.
- K. Overloop, P. Van Hecke, F. Vanstapel, H. Chen, S. Van Huffel, A. Knijn and D. van Ormondt, NMR Biomed., 1996, 9, 315 CrossRef CAS.
- H. Chen, S. Van Huffel, D. van Ormondt and R. de Beer, J. Magn. Reson. A, 1996, 119, 225 CrossRef CAS.
- J. Totz, A. van den Boogaart, S. Van Huffel, D. Graveron-Demilly, I. Dologlou, R. Heidler and D. Michel, J. Magn. Reson., 1997, 124, 400 CrossRef CAS.
- L. Vanhamme, A. van den Boogaart and S. Van Huffel, J. Magn. Reson., 1997, 129, 35 CrossRef CAS.
- L. Vanhamme, S. Van Huffel, P. Van Hecke and D. van Ormondt, J. Magn. Reson., 1999, 140, 120 CrossRef CAS.
- I. Dologlou, S. Van Huffel and D. Van Ormondt, J. Magn. Reson., 1998, 130, 238 CrossRef CAS.
- L. Vanhamme, T. Sundin, P. Van Hecke, S. Van Huffel and R. Pintelon, J. Magn. Reson., 2000, 143, 1 CrossRef CAS.
- T. Sundin, L. Vanhamme, P. Van Hecke, I. Dologlou and S. Van Huffel, J. Magn. Reson., 1999, 139, 189 CrossRef CAS.
- Z. Bi, A. P. Bruner, J. Li, K. N. Scott, Z. S. Liu, C. B. Stopka, H. W. Kim and D. C. Wilson, J. Magn. Reson., 1999, 140, 108 CrossRef CAS.
- B. Antalek and W. Windy, J. Am. Chem. Soc., 1996, 118, 10331 CrossRef CAS.
- W. Windig, J. P. Hornak and B. Antalek, J. Magn. Reson., 1998, 132, 298 CrossRef CAS.
- B. Antalek, J. P. Hornak and W. Windig, J. Magn. Reson., 1998, 132, 307 CrossRef CAS.
- W. Windig and B. Antalek, Chemom. Intell. Lab. Syst., 1999, 46, 207 CrossRef CAS.
- W. Windig, B. Antalek, L. J. Sorriero, S. Bijlsma, D. J. Louwerse and A. K. Smilde, J. Chemom., 1999, 13, 95 CrossRef.
- N. Zumbulyadis, B. Antalek, W. Windig, R. P. Scaringe, A. M. Lanzafame, T. Blanton and M. Helber, J. Am. Chem. Soc., 1999, 121, 11554 CrossRef CAS.
- W. Windig, B. Antalek, M. J. Robins, N. Zumbulyadis and C. E. Heckler, J. Chemom., 2000, 14, 213 CrossRef CAS.
- T. Brekke, O. M. Kvalheim and E. Sletten, Chemom. Intell. Lab. Syst., 1989, 7, 101 CrossRef CAS.
- J. T. W. E. Vogels, A. C. Tas, F. van den Berg and J. van der Greef, Chemom. Intell. Lab. Syst., 1993, 21, 249 CrossRef CAS.
- J. T. W. E. Vogels, A. C. Tas, J. Venekamp and J. van der Greef, J. Chemom., 1996, 10, 425 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2001 |