Electric field-induced mobilisation of multiphase solution systems based on the nitration of benzene in a micro reactor
G. N. Doku, S. J. Haswell*, T. McCreedy and G. M. Greenway
Department of Chemistry, University of Hull, Hull, UK HU6 7RX
First published on 19th December 2000
Abstract
This paper describes the electric field-induced flow characteristics of multiphase solutions in a micro reactor device using the nitration of benzene as a model process. Photolithographic and wet etching techniques were used to fabricate the micro reactor (channels, 200 μm id, 100 μm deep) in a borosilicate glass substrate. The results focus specifically on the flow parameters of reagents/reactants (i.e., voltage, solution concentration and pH ranges and current–voltage relationships) used in this study. The benzene was introduced and mobilised by electroosmotic flow (EOF), as a microemulsion using an appropriate surfactant (sodium dodecyl sulfate), whilst the nitronium ions, produced in situ from mixed H2SO4–HNO3 (the nitrating agent), underwent electrophoretic-induced (electrokinetic) mobility. A co-surfactant, butan-1-ol, was used owing to (a) its relative solubility in the aqueous surfactant solution, (b) its ability to aid the solubilization of benzene, (c) the provision of a water-rich (oil-in-water) rather than oil-rich (water-in-oil) microemulsion system and (d) its lack of significant adverse effects on the EOF. The optimum conditions used for the nitration of benzene within the micro reactor were a run of the microemulsion as main reagent stream, then three 30 s segmented injections of mixed acid, with a 5 s push of the microemulsion into the system after each injection, and then a 60 s stopped-flow reaction time before driving reaction product segments to a collection reservoir.
1. Introduction
Whereas the field of micro total analytical systems (μTAS), based on the use of electroosmotic flow (EOF) and electrophoretic separations,1–8 has gained much attention in recent years, the application of complementary micro reactor technology based on the electric field-induced mobility for chemical reactions9,10 and in particular organic synthesis11 has attracted less attention. In addition, solutions employed to date in such systems have primarily been aqueous or single solvent phases. Early work, however, involving the nitration of nitrophenol, indicated that performing multiphase reactions in micro reactors was feasible.12 Furthermore, this provisional research suggested that the position of the nitration and thus the isomers of nitrophenol produced may be influenced by the operating conditions used. In this work, the nitration of benzene using mixed acid, H2SO4–HNO3, as the nitrating agent was used as a model process to investigate the ability to move multiphase solutions in a microreactor using electrically driven flow. The benzene was introduced into the system as a microemulsion using an appropriate surfactant, as this provided the necessary ionic character to mobilise the organic system under an electric field. In addition, the formation of the microemulsion droplets of the benzene would increase the surface area available to the nitrating agent, as the rate of nitration is determined not only by the chemical kinetics of the reaction, but also by the mass transfer between the two phases.13 The choice of a surfactant–co-surfactant system was based on an evaluation of what effects they will have on both the EOF and reaction mechanism. In addition, due consideration was given to the solubilization power of the surfactant–co-surfactant and the ease with which a water-rich (oil-in-water) rather than oil-rich (water-in-oil) microemulsion system can be formed in a stable manner.2. Experimental
2.1. Reagents and materials
All reagents were of the analytical-reagent grade unless stated otherwise. The primary reagents used were disodium tetraborate (borax, 98%) (Merck, Poole, Dorset, UK), sodium dodecyl sulfate (SDS, 99%) (Lancaster Synthesis, UK), methanol (99.8%, 0.791 g ml−1) (BDH, UK), ethanol (absolute) (bottled by U.O.H), propan-1-ol (99.5%) (BDH, butan-1-ol (99%) (BDH), pentan-1-ol (98%, 0.814 g ml−1) (BDH), hexan-1-ol (98%, density 0.814 g ml−1), (Aldrich, UK), octan-1-ol (99%) (Avocado, UK), decan-1-ol (0.83 g ml−1) (BDH), benzene (99.8%, 0.878 g ml−1) (Fisons, UK), sulfuric acid (98%) (Fisher Scientific, Loughborough, Leicestershire, UK) and nitric acid (68%) (Prolabo, Fontenay s/Bois, France). High purity distilled, de-ionised water (Elgastat UHQ PS system, Elga, High Wycombe, UK) with a conductivity of 18 (mΩ cm)−1 was used. Hydrofluoric acid (40%) and ammonium fluoride were obtained from Merck (Poole, Dorset, UK). Microposit chrome etch 18 and photoresist remover 1112A were purchased from Shipley (Coventry, UK). The glass for the micro reactor chips and cover-plates were both superwhite Crown B70 borosilicate glass (Instrument Glasses, Enfield, UK).2.2. Chip device and instrumentation
The micro reactor consisted of channels 200 μm in diameter and 100 μm deep (Fig. 1), fabricated in borosilicate glass (25 mm long, 25 mm wide and 3 mm thick), using a wet-etching technique described previously.14 The glass chip was thermally bonded to a thicker glass cover-plate (25 mm long, 14 mm wide and 17 mm thick, also of borosilicate) containing 2 mm holes pre-drilled to align with the ends of channels which served as reservoirs for reagents and analyte and to support the platinum electrodes used to apply voltages to the solutions. | ||
| Fig. 1 Micro reactor design used for the nitration of benzene. BC = 3 mm, CD = DE = 4 mm, RB = SC = W2D = W1E = 5.5 mm, BE = 11 mm, RW1 22 mm, SW2 = 14 mm. | ||
A Farnell (Leeds, UK) HiVolt XRV30/1.7 reversible polarity power supply was used to supply the electric field to the system. This offered a maximum power output of 50 W with variable voltages up to 30000 V and currents up to 1.7 mA to be selected in constant stabilised current or voltage modes, with an adjustable trip for overcurrent protection. The power supply was connected via high purity copper conductors to 0.15 mm diameter platinum electrodes with a 10 mm exposed length for interfacing with the solutions in the reservoirs. For safety, the electrode system was built in an isolation box fitted with a power cut-out preventing high voltage operation when the lid was opened. The voltages applied across the electrodes and the internal currents generated within the solutions in the channels were monitored on an AVO M2036 digital autoscaling multimeter (Thurnby Thunder Instruments, Huntingdon, UK) connected in parallel with the device.
Gas chromatography-mass spectrometry (GC-MS) with a GC970870-MS200478 system (Finnigan MAT GCQ, Austin, TX, USA), was used to analyse for benzene mobilised within the channel by the bulk flow of the microemulsion, and any benzene-nitrated products. The GC column used was a 35 m × 0.25 mm id Rtx-5MS capillary (Crossbond 5% diphenyl–95% dimethylpolysiloxane stationary phase), stable to 360 °C; the carrier gas was helium at a flow rate of 40 cm3 s−1. The temperatures used were: injector 250, detector 250 and oven 85 °C (held for 3 min) ramped at 30 °C min−1 to 250 °C (held for 15 min).
2.3. Characterisation of electroosmotic flow
The main EOF characteristics monitored were applied voltage, current–voltage relationships and solution concentration and pH, which were then selected to obtain parameters for stable electric field-driven flow.The solution under investigation was placed in reservoir R (Fig. 1) and water or buffer in reservoir W1, both to pre-marked levels. The reservoir levels of other reagent solutions in the side channel reservoirs S and W2 perpendicular to the main channel R–W1 were also recorded. The positive electrode was placed in R and the negative electrode in W1; the other reservoirs were electrically left floating.
Initially, different disodium tetraborate buffer solutions of concentrations 20 mM (pH 9.25), 15 mM (pH 9.24), 10 mM (pH 9.22) and 5 mM (pH 9.11) were run at different applied voltages (200–1200 V) over a period of 15 min and the change (decrease) in volume of buffer in reservoir R was measured by recording the difference in reagent level in the reservoir. This was done by filling the reservoir to its original level after each 15 min run and recording the addition required with a graduated 100 μl HPLC syringe. The voltage (V), current (μA), time (min) and volume changes (μl) were recorded and the flow rates (μl min−1) calculated to obtain an optimum buffer concentration that could be used in conjunction with the reagents having a pH > 4.
In a similar manner, the EOF properties of SDS solutions of concentrations 100 mM (pH 6.32), 50 mM (pH 6.05), 20 mM (pH 5.75), 15 mM (pH 5.80), 10 mM (pH 5.78) and 5 mM (pH 5.47) were studied to verify the effect of voltage and/or current on the flow. In addition, it was established which concentrations of the SDS could be used as the micellating or microemulsification surfactant in the buffer solution.
The above approach was repeated to establish the flow characteristics for different SDS–borate buffer solution mixtures containing 15 mM SDS (the SDS concentration that gave the best EOF properties) at different borate buffer concentrations, 5, 10, 15 and 20 mM, giving solution mixtures of pH 9.13, 9.24, 9.26 and 9.28, respectively. Finally, the buffer concentration was kept constant at 10 mM Na2B4O7 (the buffer concentration that gave highest EOF rate) whilst the SDS concentration was varied to verify the effect of SDS concentration on the EOF of the mixture. In this case, the actual SDS concentrations and the pH of the solution mixtures were 5 mM (pH 9.06), 10 mM (pH 9.11), 15 mM (pH 9.24), 20 mM (pH 9.25), 50 mM (pH 9.28), 100 mM (pH 9.33) and 200 mM (pH 9.38).
A number of straight chain alcohols, methanol, ethanol, propan-1-ol, butan-1-ol, pentan-1-ol, 1-hexan-1-ol, octan-1-ol and decan-1-ol, were considered as possible co-surfactant agents but owing to solubility considerations only methanol to butanol were investigated. Initially, separate 10% v/v alcohol (methanol to butanol) mixtures with 10 mM borate buffer–SDS solution mixtures with various SDS concentrations (5–200 mM) were studied, to establish the effect of the presence of the alcohols on the EOF, and to establish how the SDS concentration in alcohol solution would affect the EOF. In a further study, the alcohol concentration was varied from 5 to 50% in separate 50 mM SDS–10 mM borate buffer solutions. This was followed by preparing separate 100 mM SDS–10mM borate buffer solutions to investigate the effect of alcohol concentration on the EOF and thus the possibility of using higher co-surfactant (alcohol) levels at higher surfactant (SDS) levels since this may be the only conditions under which some non-polar organic compounds would be solubilized.
Six microemulsion systems of the quaternary system, H2O–SDS–butanol–benzene, one oil-rich (water-in-oil) and five water-rich (oil-in-water) with various water contents, were prepared by selecting various relative percentage weight ratios of each of the components from a Winsor IV phase diagram.15 The actual corresponding amounts (in weight or volume) were measured and mixed with gentle agitation and allowed to settle for 4 h to give clear or nearly clear microemulsion systems. The EOF properties of the different microemulsion systems were then studied at voltages ranging from 100 to 500 V to ascertain the trend in flow and the voltage–current characteristics as the mixture moved from the oil-rich to the more water-rich microemulsion systems. This was followed by evaluating the amounts of benzene moved by EOF for the five oil-in-water microemulsion systems (the oil-rich system did not exhibit any EOF) under an applied field of 100 V, by taking a 0.5 μl aliquot of the resulting solution in the collection reservoir W1 (Fig. 1) and analysing it by GC-MS (single-ion MS monitoring at masses 76, 77, 78 and 79). A portion (0.5 μl) of the remaining solution in the reagent reservoir R was similarly analysed to determine how much benzene was left in the reagent reservoir R after applying the electrical field.
A stock standard 11.22 M H2SO4–5.37 M HNO3 solution was prepared and serial dilutions (dilution factors 1.333, 1.5, 1.666, 1.82, 2, 2.5, 3, 4, 5, …, 10) were carried out to produce other mixtures of concentrations ranging from 8.42 M H2SO4–4.03 M HNO3 to 1.22 M H2SO4–0.53 M HNO3, which were used in similar electrokinetic flow studies at voltages ranging from 100–500 V to ascertain the trend in bulk flow and the effect of the acid concentration on the current.
The optimum acid concentration for the model nitration reaction and the maximum voltage and time of field application suitable for driving the mixed acid in the system, with microemulsion (containing organic components) also present, were investigated. For practical reasons, i.e., minimising Joule heating effects and stabilising the current flow properties of the two reactants, it was necessary to select a set of compromised lower voltages than indicated in the univariate optimisation. It was therefore decided to pump the microemulsion at 90 V by EOF as the main reagent and inject the mixed acid at 200 V.
In a further experiment, microporous silica frits16 0.5 mm in length were integrated into the channels R–B, C–S and W2–D (leaving the main reagent channel B–W1 free) to control hydrostatic pressure effects caused by imbalance in liquid levels in reservoirs, and to aid EOF.
2.4. Multiphase solution mobilisation and optimum operational conditions for the model nitration reaction (nitration of benzene)
The micro reactor manifold used for the multiphase solution mobilisation, based on the model nitration experiment (Fig. 1), contained four solution reservoirs (R, W1, S and W2). A Z-mode sample injection3 was used to introduce the mixed acid whilst running the microemulsion as the main reagent. The microreactor also contained 0.5 mm long microporous silica frits16 integrated into the channels R–B, C–S and W2–D.After loading the reagent reservoir R and sample reservoir S with 10 mM borate buffer (Na2B4O7, pH 9.22), and the waste reservoirs W1 and W2 also with buffer, the electrical fields HVPS 1 and HVPS 2 (400 V in each case) were applied one after the other to prime all channels with the blank buffer. The buffer in reservoir R was then replaced with a 92.5% water microemulsion and the sample reservoir S was loaded in a similar way with 6.17 M H2SO4–2.95 M HNO3. Reservoirs W1 and W2 were loaded with the buffer. The electrical field HVPS 1 (90 V, ≈270 μA) was applied between R (+) and W1 (−) for 5 min to fill the main reaction channel R–W1 with the microemulsion. A second voltage was then applied using the HVPS 2 (200 V, ≈300 μA) across S (+) and W2 (−) for 5 min to fill the sample channel C–D completely with the mixed acid, driving any microemulsion between C and D to waste W2. The HVPS 1 (90 V, ≈270 μA) was again applied across the R (+) and W1 (−) to drive any mixed acid and buffer along the main reaction channel to W1. After replacing the contents of reservoirs W1 and W2 with fresh 10 μl of buffer, the HVPS 2 [S (+), W2 (−)] was then applied for 180 s to inject the mixed acid into the microemulsion stream and, after allowing a 60 s stopped-flow mode as initial reaction time, the HVPS 1 was then applied across R (+) and W1 (−) again for 5 min to drive the reaction segment to the reservoir W1. A 100 μl HPLC syringe, pre-loaded with 6 μl of octane, was then used to transfer the reaction mixture in W1 to a 90 μl sample glass tube, followed by introduction of a further 6 μl of octane to assist in the extraction of any reaction products into the top organic layer.
Using fresh microemulsion and fresh mixed acid solution in each case, the process was repeated with different injection times between 0 and 120 s to verify the optimum continuous sample injection time for the process, and the corresponding reaction mixtures were collected separately. In this way, the amount of microemulsion fed into the system was kept constant whilst that of the mixed acid was varied. Before using the solutions, the 10 mM borate buffer, the 6.17 M H2SO4–2.95 M HNO3 and the 92.5% water microemulsion were cooled to 4 °C in a refrigerator to minimise the temperature effects associated with the exothermic nitration reaction. At injection times >1 min, bubble generation was observed in the injection line S–W2 owing to excessive heating. In the injection process, the switching off and on of the microemulsion flow (R–W1) and the mixed acid reactant (S–W2) was performed simultaneously. Each of the mixtures produced was stirred with a small stainless steel rod, allowed to settle and 1 μl each of the octane extracted solutions was then analysed by GC–MS. In this part of the work, 0.5 μl of pure nitrobenzene and then pure octane were first injected in turn to determine the respective retention times and their MS data in full scan mode (mass 70–220).
Experiments based on a segmented mixed-acid injection coupled with a non-stopped-flow mode of operation, but with different segmented microemulsion run times, were also carried out to verify the effects of these conditions on the product yield. In these experiments, after priming all channels with the borate buffer, the main reaction line R–W1 was filled with the microemulsion, and the sample channel S–C was filled with the mixed acid, as before. Reservoirs W1 and W2 were each loaded with a fresh 10 μl of buffer. The HVPS 2 [S (+) W2 (−)] was then applied for 30 s (the optimum mixed-acid injection time) to inject the mixed-acid into the microemulsion stream and, without allowing any initial stopped-flow reaction time, the HVPS 1 [R (+) W1 (−)] was introduced for 30 s to run the microemulsion and drive the sample segment away from the injection point, followed by a further two 30 s mixed-acid injections with 30 s microemulsion runs between injections. Finally, the HVPS 1 was applied from R (+) to W1 (−) for 8 min to drive all sample segments to reservoir W1.
Using fresh microemulsion and fresh mixed acid solution in each case, the process was repeated with the same three times 30 s mixed-acid injection sequence but using different microemulsion run times of 0–25 s between injections and the corresponding reaction mixtures were collected separately. In this way, in addition to segmenting the injection and verifying the effect of non-stopped flow mode on the reaction, the amount of mixed acid introduced into the system was kept constant at three 30 s injection volumes, whilst the amount of microemulsion fed into the system was varied by altering the segmented microemulsion run times. The extraction of samples from the collection reservoir and subsequent GC–MS analysis were performed as described previously.
3. Results and discussion
3.1. EOF properties of buffer, surfactant and alcohol reagent systems
In order to establish the suitability of pumping a microemulsion in a borate buffer solution by EOF, the flow characteristics of the borate buffer were initially investigated to identify the optimum concentration required when used in conjunction with the surfactant SDS and other reagents. The anionic surfactant SDS was employed because it is known to solubilize benzene strongly,15,17–19 especially when employed with a co-surfactant such as an alcohol. In addition to providing an ionic character to the organic system, the small droplets (10 nm) produced increased the surface area available for the nitronium ion attack. SDS was also expected to exhibit an extra positive catalytic effect on the benzene reactions since it would exert an attraction on attacking electrophiles on to the surface of the droplet to aid attack on the solubilized benzene. It was expected that SDS molecules would not adsorb on the micro-channel surfaces of the microreactor, thus posing no adverse effects on the EOF, since the SDS and the glass surface are both negatively charged.The borate buffer solutions alone all gave good EOF under applied voltage over the concentration range 5–20 mM studied, with the flow rate changing very little as a function of concentration. The highest EOF of 2.6 μl min−1 was recorded for the 10 mM solution.
On the whole, the surfactant solutions alone also moved well by EOF within the voltage (200–1200 V) and concentration (5–100 mM) ranges studied, since SDS is an anionic surfactant and had no detrimental adsorption effects on the capillary surface. The 15 mM SDS solution gave the maximum flow rate of 2 μl min−1. Generally, the flow rates recorded for the surfactant solutions (pH 5–7) were lower (1.2–2 μl min−1), however, than the 2.3–2.6 μl min−1 obtained for the borate buffer solutions (pH 9.13–9.33), owing to the lower pH and the slightly viscous nature of the surfactant solutions.
In order to use the surfactant (SDS) in a borate buffer solution, it was clearly necessary to establish the EOF parameters of the surfactant–buffer solution mixtures, across a range of different relative concentrations. Indeed, the dissolution of the SDS in the borate buffer solutions was found to improve the EOF rates, over the SDS solution systems without buffer, owing mainly to the pH buffering effect (pH 9.06–9.48 range) produced by the borate buffer. It was observed, however that beyond 100 mM SDS concentration, voltages >300 V resulted in higher and fluctuating currents, causing some degree of Joule heating to occur. SDS concentrations >100 mM were therefore thought to be a practical limitation. In Fig. 2, for example, the errors in the measured flow rates of the SDS–buffer mixtures (with no alcohol present) were in the range 1–2% RSD, with the error increasing as a function of the SDS concentration, possibly owing to the current fluctuations resulting from the increasing Joule heating effects.
 | ||
| Fig. 2 Plots of flow rate versus SDS concentration for SDS–10 mM borate buffer solutions without alcohol, and flow rate versus SDS concentration for systems containing 10% v/v alcohols (methanol to butanol), at 800 V. | ||
The flow rates recorded for all the alcohol containing solution mixtures were slightly lower than those obtained for the mixtures without alcohol (Fig. 2). The presence of the alcohols resulted in a slight suppression of the EOF owing to their organic nature and possibly a slightly non-ionic surfactant adsorption property on the capillary wall. All four alcohols investigated (methanol, ethanol, propanol and butanol), however, gave nearly the same EOF properties, with butanol showing only a slight lowering of the EOF compared with the other alcohols. The errors in the measured flow rates of these alcohol containing mixtures were between 1.0 and 2.5% RSD for all four alcohols, with the error again increasing as a function of the SDS concentration. For all the four alcohols investigated, the EOF became suppressed as the alcohol concentration increased in both the 50 mM (Fig. 3) and the 100 mM SDS solutions (Fig. 4), owing to the increase in the organic character and viscosity of the solutions. In addition, it was possible that the alcohol was adsorbed on the capillary surface, so affecting the zeta potential of the system.20 The general trend observed (Fig. 3 and 4) as the alcohol concentration increased was that the flow rate of the individual alcohol systems all approached a convergence value at about 50% alcohol concentration. Beyond 40% alcohol concentration for the three most soluble alcohols (methanol, ethanol and propanol), the Joule heating and bubbling effects became more pronounced, especially at high SDS concentrations and applied voltages >600 V, owing to the higher volatility of these alcohols (methanol, ethanol and propanol). In these cases of different alcohol concentrations (Fig. 3 and 4), the errors in the flow rate measurements were between 2 and 3% RSD, with the error increasing slightly with increasing alcohol concentration. It is important to note, however, that in the SDS operating concentration range of 0–100 mM, as the concentration of alcohol or any other organic compounds increases, the SDS concentration must also be increased to ensure that sufficient ions are present in the medium to induce solution mobility under applied field.
 | ||
| Fig. 3 Plots of flow rate versus alcohol concentration in 50 mM SDS–10 mM borate buffer, at 800 V. | ||
 | ||
| Fig. 4 Plots of flow rate versus alcohol concentration in 100 mM SDS–10 mM borate buffer, at 800 V. | ||
The solubilizing properties of SDS–alcohol in a microemulsion increases substantially with increasing salinity.19 In this work, any additional salinity was expected to be provided by the sodium borate buffer employed.
3.2. EOF properties of the microemulsion systems
Of the four alcohols investigated for EOF purposes, butanol was selected because it has the highest threshold number15 of carbon atoms, nC = 4, i.e., where the unique Winsor IV domain is broadest and therefore there is more opportunity for changing the relative percentage composition of components without extending beyond the boundaries of the microemulsion domain. The percentage compositions obtained and the actual quantities of components measured to generate the microemulsion systems are shown in Table 1.| Water-rich (oil-in-water) system | ||||||
|---|---|---|---|---|---|---|
| Parameter | Oil-rich system | 1 | 2 | 3 | 4 | 5 |
| CW (%) | 32.50 | 70.00 | 79.38 | 85.00 | 88.75 | 92.50 |
| (2.6 g, 2.6 ml) | (10.18 g, 10.18 ml) | (15.88 g, 15.88 ml) | (26.7 g, 26.7 ml) | (30.43 g, 30.43 ml) | (49.33 g, 49.33 ml) | |
| CH (%) | 31.25 | 9.38 | 5.63 | 3.75 | 2.50 | 1.88 |
| (2.5 g, 2.9 ml) | (1.36 g, 1.58 ml) | (1.13 g, 1.31 ml) | (1.0 g,1.16 ml) | (0.86 g, 0.99 ml) | (1.0 g, 1.16 ml) | |
| CS (%) | 37.50 | 20.63 | 15.00 | 11.25 | 8.75 | 5.63 |
| SDS (%) | 12.50 | 6.88 | 5.00 | 3.75 | 2.92 | 1.88 |
| (1.0 g) | (1.0 g) | (1.0 g) | (1.0 g) | (1.0 g) | (1.0 g) | |
| ROH (%) | 25.0 | 13.75 | 10.00 | 7.50 | 5.83 | 3.75 |
| (2.0 g, 2.5 ml) | (2.0 g) | (2.0 g) | (2.0 g) | (2.0 g) | (2.0 g) | |
| Total volume/ml | 8.0 | 14.28 | 19.68 | 30.36 | 33.92 | 52.99 |
After mixing the components of the microemulsion and applying some agitation, clear microemulsion media were successfully obtained at room temperature. The time required for equilibration was, however, found to increase as the water content increased; thus, the oil-rich microemulsion systems 1 and 2 required between 1–4 h to become clear whereas the more water-rich systems 3, 4 and 5 required 8–12 h. In each case, the formation was found to be slightly endothermic and any foaming disappeared after the equilibration.
Fig. 5 shows the plots of the EOF rate versus voltage for the different microemulsion systems investigated for different percentage water content. The oil-rich microemulsion system 1 exhibited no bulk solution movement over the whole voltage range studied. This was thought to be owing to the high levels of SDS, alcohol and benzene components of the microemulsion which made the solution very oily (viscous) and interfered with the channel surface. With this oil-rich microemulsion system, the recorded currents were very high and fluctuated owing to high resistance, creating strong Joule heating in the system. It was found that, because the organic (benzene and butanol) additives generally decreased the vapour pressure of the whole mixture and the amount of SDS was substantial, evaporation occurred which precipitated the SDS in the reservoir, making its use impractical. The water-rich (oil-in-water) microemulsions showed a steady increase in the EOF, with a corresponding decrease in the current required. This trend was accompanied by a more stable current and less Joule heating, as the samples moved to more water-rich and lower organic/surfactant concentration systems. Voltages as low as 100 V and a current of 28 μA were found to be sufficient to move the water-rich microemulsion solutions. The three most water-rich microemulsion systems (containing 85.0, 88.75 and 92.5% water, respectively) showed similar and steady EOF properties over the voltage range studied, but with decreasing current fluctuation and Joule heating effects. The errors in the flow rate measurements were between 1 and 2% RSD, with the error decreasing as the water content of the microemulsion increased.
 | ||
| Fig. 5 Plots of flow rate versus voltage for the microemulsion systems with different percentage water contents. | ||
3.3. Flow properties of the nitrating mixed-acid solutions
Fig. 6 shows the plots of the volumetric flow rate versus applied voltage for a range of acid concentrations at a fixed H2SO4 to HNO3 ratio. The nitrating mixed-acid systems showed a steady decrease in flow rate as the concentration of the acids increased, with the highest concentration (11.22 M H2SO4–5.37 M HNO3) showing almost a zero flow rate within the micro reactor. Owing to the low pH (<2) and high ionic strength of the mixed-acid solutions, the bulk mobility observed for the less concentrated solutions was more likely to be induced by bulk ionic electrophoretic movement, rather than EOF. For the more dilute solutions with concentrations less than 5.61 M H2SO4–2.68 M HNO3, voltages as low as 100 V appeared to be sufficient to move the solutions in the micro reactor. In addition, there was reduced Joule heating owing to the low electrical resistance. The errors in the flow rate measurements for the acid solutions were between 1.5 and 2.5% RSD, with the error decreasing as the acid concentration decreased. In all cases, no evolution of brown nitrogen dioxide gas was noticed at the electrodes and in the channels.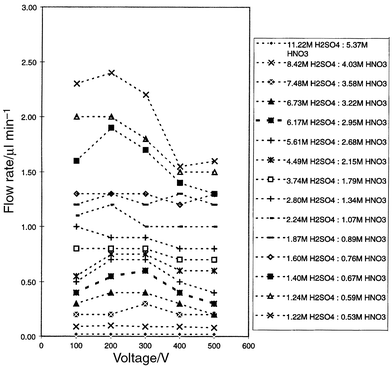 | ||
| Fig. 6 Plots of flow rate versus voltage for the mixed-acid concentration levels. | ||
Flow rates were determined by the simple volume change measurement procedure,20 avoiding current-21 and optical-based21 methods which when applied to a two-phase system may pose some significant problems. For example, the presence of organic components in the aqueous phases and the employment of solutions with pH < 2 may result in some current irregularities, which would make flow rate measurements by current–time monitoring difficult. However, the integration of UV/visible optical detectors to allow flow rate measurements by more conventional approaches through the use of injected neutral marker21 is currently under investigation.
3.4. Characteristics of the nitration model reaction based on the electric field-induced mobilisation of the multiphase system
Ideally, the yield of reaction product should be calculated relative to the amount of reagent or sample injected into the reactor channel. However, this was difficult to achieve in this investigation as the nitronium ions injected are generated as reaction intermediates in the reversible interaction between H2SO4 and HNO3. Hence, the yields of any benzene-nitrated products (as absolute gram amount and as percentage conversion) were calculated relative to a theoretical yield based on 100% conversion of the amount of benzene in an estimated volume of microemulsion interacting with the injected mixed acid at the reagent–sample interfaces initially created at the channel junction C (Fig. 1). The volume of the channel junction C was calculated to be 4 nl and the total amount of microemulsion interacting within the injected mixed acid at both sides of the injected slug was estimated to be approximately half (i.e., 2 nl) of the volume of the channel junction.22Fig. 7 shows the percentage yield (calculated as percentage mass) for mono-, di- and trinitrobenzene obtained by injecting a slug of the mixed acid, for different periods of time, from reservoir S into the microemulsion filled main channel. During this process the flow was stopped for 60 s after each injection to allow the reaction to proceed.
 | ||
| Fig. 7 Plots of the percentage yield for mono-, di- and trinitrobenzene obtained by varying the injection times of the mixed acid into the microemulsion filled main channel. | ||
The products formed were found to be dependent on the injection time and thus the corresponding volume of reactant acid introduced into the microemulsion stream. For the mono-nitrobenzene an injection slug of acid between 30 and 60 s proved to be optimal, giving a yield of 62–65%, but the less abundant di- and trinitrobenzene species increased slowly with a corresponding increase in acid injection time. It is important to note that whereas EOF, with a characteristic flat (trapezoidal or rectangular) flow profile, is the main mechanism for solution mobility, electrophoretic mobility will also occur, affecting the diffusion limiting reactions taking place in the interfacial zones between the reactants.22 The amount of reaction product formed would therefore depend not only on the volume injected but also on the diffusion constants for the solutions present, the individual electrophoretic mobility for each component and the reactant concentration. Thus, if insufficient acid is injected the nitrobenzene yield will be reduced (i.e., <30 s). Attempts to deliver more acid by increasing the injection time (i.e., >60 s) had a limiting effect on the amount of product obtained and this trend could possibly be due to an increase in Joule heating effects creating some in-channel bubbling during the injection, which eventually stopped the fluid flow. To prevent such excessive time-dependent Joule heating effects, a 30 s injection time, coupled with the 60 s initial stopped-flow mode reaction time, were chosen. This corresponds to <5 nl injected mixed-acid volume completely filling the reagent-sample channel junction C, and about 2 nl of microemulsion interacting with the acid at the interfaces between the microemulsion and the mixed-acid. To establish fully the optimum reaction conditions, experiments were carried out in which the mixed acid was segmented by different volumes of microemulsion, operating in a continuous flow mode. These results showed that the most appropriate injection sequence for this reaction was three 30 s segmented mixed-acid injections each immediately followed by a 5 s flush of microemulsion into the system. Clearly, however, the whole aspect of sample injection and reaction characteristics in μFIA systems requires a more detailed study, which is currently in progress.
It is also important to stress that in the absence of any turbulent mixing, to disperse the injected mixed-acid slug in the relatively larger volume of microemulsion in the channel means that some concentrated mixed acid would still be in direct interfacial contact with the already formed nitrobenzene product. This situation could therefore lead to the possible formation of polynitration products. However, only traces of the di- and trinitrobenzene were formed owing to the deactivating nature of the nitro group already present on the benzene ring and the limited amount of mixed acid introduced as a sample into the benzene stream. Thus, kinetically, the formation of the polynitrated products was found to be, as expected, a function of both time and the amount of mixed acid introduced into the system. Shorter reaction times and less acid (or excess microemulsion) would favour the production of the mononitrobenzene, whereas a longer time and excess acid would favour the production of the polynitrated products.
Generally, any temperature gradients within the channels, resulting from the exothermic nature of the reaction and Joule heating effects, are expected to be minimal owing to the relatively small volume present. Locally high thermal effects may, however, still occur and the absence of active cooling may influence the reaction equilibrium to some extent, leading to decomposition of the reactants. In addition, the displacement of some of the surfactant SDS as sulfonic acid and the borate buffer as boric acid by the H2SO4–HNO3, together with the presence of water and other side-reactions consuming some free hydrogen ions in solution, will act to suppress the generation of the electrophilic nitronium ion. As a consequence, the yields are expected to remain lower than that of the conventional industrial process. Other contributing factors to these low yields could be attributed to the product collection from the collection reservoir and the extraction efficiency of octane.
4. Conclusions
This investigation has demonstrated that it is possible to mobilise a non-polar liquid (benzene) under EOF by dissolving it in an oil-in-water microemulsion. It should be pointed out, however, that other methods such as the addition of ion pairs (e.g., a quaternary ammonium salt)12 and electrohydrodynamics23 are possible options for mobilising relatively non-polar compounds. It was also possible to mobilise concentrated acid solutions by bulk electrokinetic mobility of solution ions at low pH.The optimum conditions for the nitration of benzene in the microreactor developed were a run of the microemulsion as main reagent stream, then three 30 s segmented mixed-acid injections each immediately followed by a 5 s flush of microemulsion into the system. This was followed by a 60 s stopped-flow reaction time before driving all reaction product segments to the collection reservoir. An in-channel microporous silica frit integrated in the manifold system was found to be helpful in reducing hydrodynamic effects, so offering greater control over the fluidics.
Clearly, it is possible to use time, applied electric field and relative amount (feed rate) of reacting species (acid or microemulsion) to control the relative yields of the various products as required. The most important factors leading to the production of the mononitrobenzene were higher rates of microemulsion feed into the system, lower acid feed rate and shorter reaction times (60 s maximum). High interaction between the levels of SDS and the percentage of sulfuric or nitric acid (in the nitrating mixture) used were also found to influence the production of mononitrobenzene. Lower amounts of the di- and trinitrobenzene were found to occur owing to the deactivating nature of the nitro groups already present on the benzene ring and the limited amount of mixed acid introduced as a sample into the benzene stream serving as the main reagent.
The synthesis of the nitrated benzene compounds using such microtechnology indicated that the reaction itself was feasible on a small scale. The system operational variables such as the applied voltage and current, reagent concentrations and pH, relative flow rates of reactant species into the system, channel sizes, etc., which could influence chemical reactions on this small scale are important considerations in the manifold design if simple systems are to be realised. Control over the selectivity of reaction products, i.e., mono-, di- and trinitrobenzene, was not conclusive. The novel mixing characteristics and the high heat dissipating capability of the glass devices together with the direct close contact of reacting species in the channels in view of the system miniaturisation could maximise the product yield.
5. References
- A. Manz, N. Graber and H. M. Widmer, Sens. Actuators B, 1990, 1, 244 CrossRef.
- A. Manz, E. J. M. Verporte, C. S. Effenhauser, N. Burggraf, D. E. Raymond, D. J. Harrison and H. M. Widmer, J. High Resolut. Chromatogr, 1993, 16, 433 CAS.
- S. J. Haswell, Analyst, 1997, 122, 1R RSC.
- Z. Fan and D. J. Harrison, Anal. Chem., 1994, 66, 177 CrossRef CAS.
- S. C. Jacobson, L. B. Kountny, R. Herenroder, A. W. Moore, Jr. and J. M. Ramsey, Anal. Chem., 1994, 66, 3472 CrossRef CAS.
- D. E. Raymond, A. Manz and H. M. Widmer, Anal. Chem., 1994, 66, 2858 CrossRef CAS.
- A. T. Woolley and R. A. Mathies, Anal. Chem., 1995, 67, 3676 CrossRef CAS.
- D. Schmalzing, L. Koutny, A. Adourian, P. Belgrader, P. Matsudaira and D. Ehrlich, Proc. Natl. Acad. Sci. USA, 1998, 94, 10273 CrossRef.
- S. J. Haswell and V. Skelton, Trends Anal. Chem., 2000, 19, 389 CrossRef CAS.
- P. D. Fletcher and S. J. Haswell, Chem. Br., 1999, 38 Search PubMed.
- G. M. Greenway, S. J. Haswell, S. O. Morgan, V. Skelton and P. Styring, Sens. Actuators B, 2000, 63, 153 CrossRef.
- S. Bird, MSc Thesis, University of Hull, 1996..
- L. F. Albright and C. Hanson, Industrial and Laboratory Nitrations, American Chemical Society, Washington, DC, 1976, p. 1. Search PubMed.
- R. N. C. Daykin and S. J. Haswell, Anal. Chim. Acta, 1995, 313, 115 CrossRef CAS.
- K. L. Mittal and B. Lindman, Surfactants in Solution, Plenum Press, 1984, vol. 3, p. 1; Search PubMed; K. L. Mittal and B. Lindman, Surfactants in Solution, Plenum Press, 1984, vol. 3, 1590. Search PubMed.
- P. D. Christenson, S. W. P. Johnson, T. McCreedy and V. Skelton and N. G. Wilson, Anal. Commun., 1998, 35, 34 Search PubMed.
- P. J. Jobe, V. C. Reinsborough and P. J. White, Can. J. Chem., 1982, 60, 279.
- R. J. Hunter, Introduction to Modern Colloidal Science, Oxford Science, Oxford, 2nd edn., 1993, pp. 1–31; Search PubMed; R. J. Hunter, Introduction to Modern Colloidal Science, Oxford Science, Oxford, 2nd edn., 1993, p. 227. Search PubMed.
- H. Kunieda and R. Aoki, Langmuir, 1996, 12, 5796 CrossRef CAS.
- G. N. Doku and S. J. Haswell, Anal. Chim. Acta, 1999, 382, 1 CrossRef CAS.
- J. E. Sandoval, Anal. Chem., 1996, 68, 2771 CrossRef CAS.
- P. D. I. Fletcher, S. J. Haswell and V. N. Paunov, Analyst, 1999, 124, 1273 RSC.
- S. E. McBride, R. M. Moroney and W. Chiang, in Proceedings of the Micro Total Analysis Sytems ’98 Workshop, ed. D. J. Harrison and A. van den Berg, Banff, Canada, October, 1998, p. 45. Search PubMed.
| This journal is © The Royal Society of Chemistry 2001 |