Gas chromatographic determination of pesticides in vegetable samples by sequential positive and negative chemical ionization and tandem mass spectrometric fragmentation using an ion trap analyser
María Dolores Hernandoa, Ana Agüeraa, Amadeo R. Fernández-Alba*a, Luis Piedraa and Mariano Contrerasb
aPesticide Residue Research Group, University of Almería, 04071, Almería, Spain.. E-mail: amadeo@ualm.es; Fax: +34 950 01 54 83; Tel: +34 950 01 50 34
bLaboratorio de Análisis Agrícolas de
COEXPHAL, Cosecheros-Exportadores de Hortalizas de Almería, 04070, Almería, Spain
First published on 12th December 2000
Abstract
A selective and sensitive chromatographic method is described for the determination of nine organochlorine and organophosphorus pesticides in vegetable samples by gas chromatography–mass spectrometry. The proposed method combines the use of positive and negative chemical ionisation and tandem mass spectrometric fragmentation, resulting in a significant increase in selectivity and allowing the simultaneous confirmation and quantification of trace levels of pesticides in complex vegetable matrices. Parameters relative to ionisation and fragmentation processes were optimised to obtain maximum sensitivity. Repeatability and reproducibility studies yielded relative standard deviations lower than 25% in all cases. Identification criteria, such as retention time and relative abundance of characteristic product ions, were also evaluated in order to guarantee the correct identification of the target compounds. The method was applied to real vegetable samples to demonstrate its use in routine analysis.
Introduction
The preservation of the environment and human health from exposure to organic contaminants is nowadays a priority objective in the European Union. In this sense, pesticides constitute a very important group of target compounds owing to their high toxicity and their unavoidable use in agricultural practice,1 in order to maintain adequate productivity in particular in agricultural areas in southern Europe.The European Community, which harmonises the registration and tolerances of pesticides throughout the Community, has established increasingly restrictive legislation relative to the maximum residue limits (MRLs) allowed in fruits and vegetables. Nowadays, many matrix–pesticide combinations are reported with MRLs between 0.01 and 0.05 mg kg−1, which represent an appreciable diminution with respect to previous limits. Such is the case with methamidophos in pepper, for which the MRL has recently been reduced to 0.01 mg kg−1 (Directive 98/82/CE). These laws and regulations have affected the limits of detection (LODs) at which analytical methods must be valid. Multi-residue methods (MRMs) applied have to be able to detect pesticides at concentrations 5–10 times below their tolerance levels, i.e. ppb or low ppb levels, if we want to guarantee the accurate identification and quantification of these residue levels.
Classical detectors currently used in the determination of organophosphorus pesticides, such as nitrogen–phosphorus and flame photometric detectors, often provide low enough LODs as a consequence of their high selectivity. The use of the electron-capture detector (ECD) also provides a very sensitive response in the determination of halogenated pesticides, but the, ECD also responds to other portions of organic molecules, which have a high electron affinity, and for that reason extensive and time-consuming clean-up procedures are necessary.
In any case, the use of these detection systems often does not meet the identification criteria required. A complete multi-residue analysis, including determination and confirmation, requires successive injections with different selective detectors and with different separation conditions, so increasing the total time of analysis. As an alternative, gas chromatography–mass spectrometry (GC-MS) has gained in popularity in recent years, because it offers simultaneous confirmation and quantification of a large number of pesticides.2–5 The main limitation lies in the relatively low sensitivity obtained for many pesticides in the full-scan mode when electron ionization (EI) is selected. The introduction of the ion trap detector (ITD) enhanced the LODs but the presence of matrix components in the samples still interferes with the results of the analysis, leading to false positive and/or false negative detection. To overcome these difficulties, it is of great interest to have more selective techniques in order to satisfy all regulatory requirements. In this sense, alternative modes of operation, such as selected ion monitoring (SIM),2 chemical ionisation (CI)6 or tandem mass spectrometry (MS-MS)7,8 have provided good and elegant alternatives. However, to our knowledge, only very few studies have been published in which these techniques were applied to vegetable samples.
In this work, a chromatographic method was developed that combines CI and MS-MS fragmentation with external source ion trap mass spectrometry (ITMS) for the determination of six organochlorine and three organophosphorus pesticides commonly used in crop protection (Fig. 1). The ability of the system to change the ionisation mode from positive (PCI) to negative (NCI) during the analysis allows the most sensitive conditions to be selected for each compound. The higher selectivity provided with CI and the lower fragmentation observed in the spectra gave rise to more abundant base peaks. These peaks were selected as precursor ions for further fragmentation by MS-MS. The product ion spectra obtained generally show sufficient fragmentation for confirmation purposes.
 | ||
| Fig. 1 Structures of the pesticides studied. | ||
The developed method was applied to the analysis of 15 vegetable samples from the COEXPHAL (Association of Producers and Exporters of Fruits and Vegetables of Almería, Andalucia, Spain) monitoring programme.
Experimental
Reagents
Pesticide standards (purity >98.0%) were purchased from Riedel-de Haën (Seelze, Germany). Individual stock standard solutions (100 mg l−1) were prepared in ethyl acetate and stored in the dark at −20 °C. Working standard solutions in ethyl acetate, containing 10 μg ml−1 of each pesticide, were used for spiking samples. Calibration standards were prepared in both ethyl acetate solution and blank extracts of vegetables. In the last case, 200 μl aliquots of the extract were evaporated to dryness under a gentle stream of nitrogen and the residue was dissolved, with sonication, in 200 μl of ethyl acetate containing the pesticides studied.Pesticide grade ethyl acetate was obtained from Merck (Darmstadt, Germany). Helium (99.999% purity) used as the carrier gas and nitrogen (99.999% purity) used for drying were purchased from Air Liquide (Madrid, Spain).
Apparatus and equipment
All the experiments were performed on a TRACE 2000 gas chromatograph (CE Instrument, Austin, TX, USA) interfaced to a GCQ ion trap mass spectrometer (Finnigan, Austin, TX, USA) equipped with external ionization, positive and negative CI and MS-MS capabilities. The analytes were separated with a Hewlett-Packard (Palo Alto, CA, USA) HP-5MS system with a cross-linked 5% diphenyl–95% dimethylsiloxane capillary column (30 m × 0.25 mm id, 0.25 μm film thickness). A 2.5 m × 0.25 mm id uncoated retention gap was coupled to the front of the analytical column, via a press fit connector. The temperature programme consisted of a 1.0 min hold at 70 °C, ramps at 35 °C min−1 to 180 °C, 10 °C min−1 to 240 °C and a final hold for 5 min. The helium flow rate was maintained at 1 ml min−1. A split–splitless injector was used in the splitless with surge mode. The injection volume was 2 μl, the injector temperature 250 °C, the surge pressure 207 kPa for 1.00 min, the split flow 50.0 ml min−1 and the splitless time 1.00 min. The transfer line temperature was set at 275 °C.Typical ITMS operating conditions were optimised by the software at the following values: electron multiplier at 1125 V, trap offset at 7 V, lens 1 at 35 V, lens 3 at 23 V and gate lens at −108 V. The external ion source was operated in the CI mode (methane as reagent gas) at 170 °C. The source pressure was optimised at 1.5 × 10−4 Torr. MS-MS conditions such as isolation (wideband application for clusters, isolation time) and excitation (resonance excitation voltage, excitation time) were optimised for the individual analytes and the results are given in Table 2.
| Wide band ion isolationa | |||||
|---|---|---|---|---|---|
| Pesticide | Chlorine atoms in parent molecule | Central value | Cluster width | Resonance excitation rf voltage (mV)b | Product ions: m/z (RA, %)c |
| a Isolation time 10 ms in all cases.b Excitation time 15 ms.c Quantification masses in bold. | |||||
| Diazinon | — | 169 | 2 | 900 | 95 (100), 141 (50), 169 (28) |
| Chlorothalonil | 4 | 266 | 6 | 1500 | 229 (100), 231 (80), 266 (28) |
| Vinclozoline | 2 | 241 | 4 | 1200 | 205 (100), 207 (90), 160 (70), 241 (15) |
| Pyrimiphos methyl | — | 306 | 2 | 1100 | 164 (100), 278 (53), 274 (49), 306 (20) |
| Chlorpyriphos | 3 | 313 | 6 | 1100 | 277 (100), 189 (70), 124 (58), 313 (17) |
| Procymidone | 2 | 283 | 4 | 1300 | 161 (100), 163 (60), 283 (28), 285 (16) |
| Endosulfan I | 6 | 270 | 8 | 1200 | 232 (100), 234 (90), 240 (19), 270 (15) |
| Endosulfan II | 6 | 406 | 8 | 1300 | 370 (100), 270 (60), 232 (50), 406 (15) |
| Endosulfan sulfate | 6 | 386 | 8 | 1600 | 351 (100), 349 (55), 353 (27), 386 (16) |
Samples
Samples of the different matrices used in this study were provided by COEXPHAL. Pesticide-free vegetables controlled by the Residue Control Laboratory of COEXPHAL were used as blank and fortified samples for recovery assays and to prepare matrix matched standards for calibration.Extraction procedure and analysis
A previously reported multi-residue extraction method2 was applied to the samples. In this method, 15 g of sample are extracted for 30 s in a Polytron with a mixture of 60 ml of ethyl acetate and 13 g of anhydrous Na2SO4. After separation of the supernatant liquid, the sample is extracted with 30 ml of ethyl acetate. A 30 ml aliquot of the combined extracts is evaporated to dryness on a vacuum rotary evaporator using a 60 °C water-bath. The residue is dissolved in 5 ml of ethyl acetate and the extract so obtained, containing 1 g of sample per millilitre of extract, is ready for GC-MS analysis.Validation of the chromatographic method
Pesticide-free vegetables controlled by the Residue Control Laboratory of COEXPHAL were used as blank and fortified samples for validation procedures. The linear relationship was studied in the range 0.005–0.10 mg kg−1 with standard solutions prepared in both blank matrix extract and ethyl acetate solvent, for comparison purposes. The repeatability of the chromatographic method was assessed by the analysis, on the same day, of five pepper extracts fortified at 10 μg kg−1 and 0.10 mg kg−1 concentration levels. Reproducibility was evaluated by the analysis of spiked samples at 10 μg kg−1 during 5 d. The LODs were determined as the analyte concentration that gave an S/N of 3, as calculated by the instrument software, and empirically verified by analysing pesticide mixtures at these concentration levels in matrix extracts.In the application of the proposed method to real samples, calibration curves generated from the standards in blank sample extracts were used for quantification. In this way, possible quantification errors originated by the matrix effect were eliminated. Integrated peak area data for 2–4 selected masses were used to construct the curves. The presence of at least four diagnostic ions at the correct retention time (<10 s) and in the correct abundance ratio was used as the identification criterion.
Results and discussion
Selection of the chromatographic conditions
The application of the chromatographic conditions proposed in the Experimental section yielded an adequate separation of the analytes studied, as can be seen in Fig. 2, which shows a full scan total ion gas chromatogram corresponding to the quantification masses (pesticides spiking level 0.01 mg kg−1). The pesticides studied and their retention times are given in Table 1. The ionisation and fragmentation conditions used in the analysis were optimised as follows.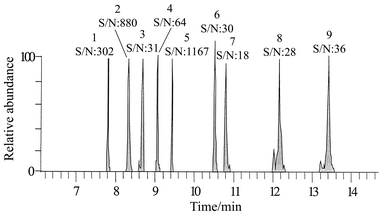 | ||
| Fig. 2 MS-MS full scan chromatogram relating to the selected quantification masses of an extract of pepper sample spiked, at 0.01 mg kg−1, with (1) diazinon, (2) chlorothalonil, (3) vinclozoline, (4) pyrimiphos methyl, (5) chlorpyriphos, (6) procymidone, (7) endosulfan I, (8) endosulfan II and (9) endosulfan sulfate. | ||
| Pesticide | tR/min | MW | Ionization mode | Main fragments, m/z (RA, %) |
|---|---|---|---|---|
| Diazinon | 7.81 | 304 | NCI | 169 (100), 303 (4) |
| Chlorothalonil | 8.32 | 264 | NCI | 266 (100), 268 (42), 264 (15) |
| Vinclozoline | 8.69 | 285 | NCI | 241 (100), 243 (63) |
| Pyrimiphos methyl | 9.05 | 305 | PCI | 306 (100), 334 (30), 346 (8) |
| Chlorpyriphos | 9.43 | 349 | NCI | 313 (100), 315 (80), 214 (7) |
| Procymidone | 10.37 | 283 | NCI | 283 (100), 285 (70) |
| Endosulfan I | 10.80 | 404 | NCI | 406 (100), 237(50), 370 (42), 270 (30), 300(36), 336(25) |
| Endosulfan II | 12.15 | 404 | NCI | 406 (100), 336 (30), 370 (24), 234 (20), 270 (15), 304 (14) |
| Endosulfan sulfate | 13.40 | 420 | NCI | 98 (100), 386 (80), 422 (20) |
Once the best ionisation mode had been selected, it was necessary to optimise the CI parameters that specially affect the sensitivity, such as source pressure and source temperature. The effect of these parameters on the sensitivity was evaluated by monitoring the peak area and S/N for the studied compounds under the different conditions assayed.
The source pressure was evaluated from 0.5 × 10−4 to 5.0 × 10−4 Torr. Optimum conditions were reached for the different compounds between 1.0 × 10−4 and 2.0 × 10−4 Torr. Therefore, as a compromise solution for all the compounds studied, 1.5 × 10−4 Torr was selected as the working pressure. In the case of the source temperature, values from 150 to 250 °C were evaluated. At higher temperatures an important decrease in the sensitivity (5–10 times lower) was observed together with an increase in fragmentation. These effects were especially intense in the case of endosulfan (I, II and sulfate). As an example, endosulfan II showed a base peak corresponding to the molecular isotopic cluster at a source temperature of 170 °C, but when the temperature was increased to 250 °C, the relative abundance of this peak decreased below 10% and abundant fragmentation occurred (base peak at m/z 270). A source temperature of 170 °C was chosen as the optimum.
Under the optimised conditions, the NCI mass spectra of most compounds exhibited 1–3 fragment ions, except endosulfan I and II, which showed more abundant fragmentation. The main fragment ions obtained and their relative abundances are given in Table 1. In the case of chlorothalonil and procymidone, no fragmentation was observed in the spectra and only the isotopic cluster corresponding to the molecular ion was present as the base peak. The molecular cluster was also present in endosulfan I and II, as the base peak, and in endosulfan sulfate, but at lower relative abundance (see Table 1), the fragment at m/z 98 being the most abundant in this case. Characteristic fragment ions at [M − Cl]− and [M − H − Cl]− have been detected in the spectra of some chlorine containing compounds (chlorpyriphos and endosulfan I , II and sulfate).
In the PCI mass spectrum of pyrimiphos methyl, the [M + H]+ ion appeared as the base peak and [M + C2H5]+ and [M + C3H5]+ as adduct ions. Therefore, little fragmentation was observed.
The optimisation of the isolation step involves, for the operator, the selection of the parent ion, the ion isolation window and the isolation time. The instrument software automatically determines other parameters associated with precursor ion isolation, such as ion rf voltage and isolation waveform characteristics (amplitudes and frequencies). The higher selectivity provided with CI and the lower fragmentation observed in the spectra gave rise to more abundant base peaks. These peaks were selected as precursor ions for further fragmentation by MS-MS in all cases, in order to obtain the largest response. In the case of endosulfan sulfate, the base peak was not selected as the parent ion because of its low mass at m/z 98. MS-MS fragmentation of this low mass ion fragment provide a higher background and insufficient structural information for identification purposes, so in this case a less abundant ion at high mass (cluster at m/z 386) was selected. The ion isolation window was selected in order to isolate the isotopic cluster for chlorinated compounds (up to ±4 units over the cluster central value). This selection permits the detection of the isotopic cluster related to the product ions and increases the qualitative information provided by the MS-MS spectra.9 The ion isolation window selected for the different analytes is given in Table 2. The isolation time was set at 10 ms in all cases.
After selecting the precursor ion for each compound and establishing adequate isolation conditions, the collision induced dissociation step of MS-MS had to be optimised. The main parameters involve in this process are the resonance excitation voltage and the excitation time. For this purpose, the GC run was divided into segments. Multiple scan events could be defined for each segment of time, within a total scan time not greater than 1.5 s. In this way, different resonance excitation voltage values could be programmed and tested during a run, so reducing the number of injections necessary for the optimisation. The final values used in this work are given in Table 2. The excitation times were kept constant at 15 ms.
After the fragmentation, characteristic fragment ions obtained were stored and sequentially ejected and detected. The MS-MS spectra obtained, under the optimised conditions, showed abundant fragmentation, sufficient for adequate identification, as shown in Table 2 where the main ions and their relative intensities are given. The parent ions were not completely dissociated, but their relative abundance was reduced to values lower than 30%. It is important to note that these data were obtained from samples containing the pesticides, at a concentration level of 100 ppb, because we observed considerable fluctuations in the relative signal intensities with concentration.
Calibration curves, repeatability and LODs
The presence of matrix interferences in sample extracts can adversely affect analyte quantification and identification. These interferences can be minimised by the application of more selective approaches such as CI or MS-MS. Nevertheless calibration curves were constructed using standard solutions prepared in both blank matrix extract and ethyl acetate solvent in order to evaluate matrix interferences, in the concentration range 5–100 μg l−1. Integrated peak area data for 1–3 selected quantification masses (shown in bold in Table 2) were used to construct the plots. Two aspects of the results were notable. First, the calibration curves obtained did not show, in general, good linear relationships in the concentration range 5–100 μg l−1. Only in the case of procymidone, vinclozoline and pyrimiphos methyl were the correlation coefficients obtained higher than 0.993. The rest of the compounds showed calibration curves which better fit a polynomial trend, and where two linear segments could be distinguished depending on the concentration range considered (see Fig. 3 for chlorpyriphos). Equations and correlation coefficients for linear and polynomial trendlines are given in Table 3. | ||
| Fig. 3 Calibration plots of chlorpyriphos in ethyl acetate (◆) and in sample extract (■). Equations and correlation coefficients for linear and polynomial trend lines. | ||
| Calibration curves (0.005–0.1 mg kg−1)a | Repeatability (n = 5) RSD (%) | |||||
|---|---|---|---|---|---|---|
| Pesticides | Linear trend (correlation coefficient) | Polynomial trend (correlation coefficient) | 0.01 mg kg−1 | 0.10 mg kg−1 | Reproducibility (n = 5) RSD (%) | Limit of detection/ μg kg−1b |
| a Equations: y = peak area and x = concentration in μg kg−1.b Corresponding to S/N 3. | ||||||
| Diazinon | y = 94.808x + 748.29 (0.978) | y = −0.42x2 + 138.98x + 18.4 (0.992) | 11 | 6 | 10 | 0.01 |
| Chlorothalonil | y = 981.4x + 9037.4 (0.957) | y = −7.23x2 + 1731.2x − 538.7 (0.992) | 6 | 5 | 11 | 0.01 |
| Vinclozoline | y = 9.8538x + 10.736 (0.995) | y = −0.01x2 + 11.04x − 4.41 (0.996) | 3 | 2 | 9 | 1 |
| Pyrimiphos methyl | y = 186.53x − 553.63 (0.997) | y = 0.25x2 + 157.6x − 53.3 (0.999) | 20 | 18 | 25 | 1 |
| Chlorpyriphos | y = 1002.3x + 1331.8 (0.978) | y = −5.91x2 + 1658.3x − 10566 (0.999) | 3 | 2 | 8 | 0.005 |
| Procymidone | y = 30.856x + 71.519 (0.993) | y = −0.09x2 + 40.3x − 49.9 (0.999) | 17 | 4 | 18 | 2.5 |
| Endosulfan I | y = 17.891x + 401.07 (0.933) | y = −0.14x2 + 32.7x + 210.8 (0.974) | 3 | 4 | 16 | 5 |
| Endosulfan II | y = 43.381x + 455.79 (0.980) | y = −0.17x2 + 61.7x + 220.9 (0.991) | 9 | 8 | 20 | 1 |
| Endosulfan sulfate | y = 20.116x + 498.84 (0.952) | y = −0.13x2 + 35.02x + 279.8 (0.989) | 4 | 3 | 13 | 5 |
Another aspect that was considered was the presence of an important matrix effect. Curves obtained with calibration standards in matrix extracts showed slopes 2–3 times higher than those obtained in ethyl acetate solvent. This enhancement of the signal in the presence of matrix can be explained by the presence of free sites in the glass injector liner, which can be occupied by matrix molecules, as previous workers have pointed out.10,11 As a consequence of these two previous observations, linear calibration curves in matrix extract were used for quantification purposes, using different sets of calibration standards depending on the concentration of the analytes in the samples.
The repeatability and reproducibility of the chromatographic method were studied by the injection (n = 5) of sample extracts spiked with the pesticides at two concentration levels (10 and 100 μg kg−1). Table 3 gives the results obtained. The repeatability of the response, at the higher concentration, was in the range 2–8% except for pyrimiphos methyl (18%). At the lower concentration, the relative standard deviations (RSDs) obtained were in a similar range, 3–11%, except for pyrimiphos methyl (20%) and procymidone (17%). The reproducibility showed RSDs between 10–25% in all cases. Overall, these values indicate a good performance of the method.
The LODs obtained for the pesticides are also given in Table 3. They were obtained by using matrix extracts in order to obtain more reliable results. In all cases, the LODs were 2–2000 times below the most stringent regulatory level established for any pesticide in the EU (0.01 mg kg−1). This guarantees the correct identification and quantification of these pesticides at these low levels.
Evaluation of identification criteria
Owing to the importance of a correct identification in order to avoid false positive detection, the criteria used for this purpose, such as retention time, presence of the precursor ions and products, selected ions (1–3) in a correct relative abundance were evaluated. No appreciable variability was observed in the retention times, despite the manual injection, showing RSDs (n = 5) <0.06%.The repeatability of the relative abundance (RA) studied at high concentration (0.10 mg kg−1) and at lower concentration was good with RSDs of 5–12% for some pesticides (diazinon, chlorothalonil, vinclozoline, pyrimiphos-methyl and procymidone). However, greater, variations were observed with endosulfan I, II and sulfate and chlorpyriphos, with RSDs of 20–30%.
Application to real samples
The applicability of the method was tested on real vegetable samples, coming from the COEXPHAL monitoring programme, and processed according to the procedure described in the Experimental section. Fig. 4 shows the MS-MS selected ion monitoring chromatograms of a real pepper sample where chlorothalonil and procymidone have been identified, at concentrations of 0.21 and 145 μg kg−1 respectively. The inset represents the MS-MS spectrum of these compounds in the sample. Good agreement of the relative abundances of the diagnostic peaks was observed between this spectrum and that obtained with the calibration solutions, thus confirming the correct identification of this compound. Other pesticides, such as vinclozoline, endosulfan and pyrimiphos methyl, were also detected in the samples. | ||
| Fig. 4 Chromatograms and MS-MS spectra of a real pepper sample where chlorothalonil and procymidone were confirmed at concentrations of 0.21 and 145 μg kg−1, respectively. | ||
Conclusions
The GC method developed in this work combines two highly selective operation modes: CI and MS/MS fragmentation. CI is a very selective technique that provides intense fragment ions but, in general, scarce structural information. This problem is overcome by the further fragmentation of an isolated precursor ion, that produces characteristic product ion spectra, sufficient for confirmation purposes. As a result, this method allowed the simultaneous quantification and confirmation of a group of nine pesticides in a complex vegetable matrix at trace levels (LODs down to 5 μg l−1 in all cases). The method fulfils the stringent EU legislative requirements with regard to precision.The effectiveness of the proposed method was evaluated by its application to real samples, demonstrating its applicability to routine analysis.
Acknowledgements
This work has been partially supported by CICYT Project [AMB98-0913-(03-03)]. The authors are grateful to ThermoQuest for instrument support.References
- D. Barcelo and M.-C. Hennion, Determination of Pesticides and Their Degradation Products in Water, Elsevier, Amsterdam, 1993. Search PubMed.
- A. Agüera, L. Piedra, M. D. Hernando, A. R. Fernandez-Alba and M. Contreras, Analyst, 2000, 125, 1397 RSC.
- J. Fillion, R. Hindle, M. Lacroix and J. Selwyn, J. AOAC Int., 1995, 78, 1252 Search PubMed.
- S. J. Lehotay and K. I. Eller, J. AOAC Int., 1995, 78, 821 Search PubMed.
- L. G. M. T. Tuinstra, A. H. Roos, A. M. Matser, W. A. Traag and J. A. van Rhijn, Fresenius’ J. Anal. Chem., 1991, 339, 384 CrossRef CAS.
- H.-J. Stan and G. Kellner, Biomed. Mass Spectrom., 1989, 18, 645 Search PubMed.
- R. J. C. A. Steen, J. L. Freriks, W. P. Cofino and U. A. Th. Brinkman, Anal. Chim. Acta, 1997, 353, 153 CrossRef CAS.
- F. J. Arrebola, J. L. Martínez-Vidal, A. Fernández Gutiérrez and M. H. Akhtar, Anal. Chim. Acta, 1999, 401, 45 CrossRef CAS.
- I. Moret, R. Piazza, A. Gambaro, P. Traldi and F. Guidugli, J. Mass. Spectrom., 1999, 34, 1389 CrossRef.
- J. Hajslova, K. Holadova, V. Kocourek, J. Povostka, M. Godula, P. Cuhra and M. Kempny, J. Chromatogr., A, 1998, 800, 283 CrossRef CAS.
- D. R. Erney, A. M. Guillespie, D. M. Giluydis and C. F. Poole, J. Chromatogr., A, 1993, 638, 57 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2001 |