Fluorimetric determination of isatin in human urine and serum by liquid chromatography postcolumn photoirradiation
Ken-ichi Mawatari*, Makiko Segawa, Rumiko Masatsuka, Yasuko Hanawa, Fumio Iinuma and Mitsuo Watanabe
Department of Analytical Chemistry, Faculty of Pharmaceutical Sciences, Teikyo University, Sagamiko, Kanagawa, 199-0195, Japan
First published on 23rd November 2001
Abstract
For the fluorimetric determination of isatin in human urine and serum, HPLC-postcolumn photoirradiation using a mobile phase has been developed. Isatin in the urine or serum sample was separated on a Capcell Pak C1 column (250 × 4.6 mm id). The mobile phase consisted of 70 mmol l−1 phosphate buffer (pH 7.2)–tetrahydrofuran (85 + 15% v/v) containing 5 mmol l−1 hydrogen peroxide, which was irradiated with germicidal light to induce fluorescence (λex 302 nm, λem 418 nm). The addition of tetrahydrofuran to the mobile phase led to the peaks showing good separation as well as increased sensitivity. The calibration graph for isatin was linear over the range of 0.16–10.7 ng. The pretreatment of the acidified urine or serum samples consisted of diluting steps or deproteinizing steps using perchloric acid, respectively. The mean recovery of isatin from urine and serum was greater than 94%.
Introduction
Isatin (indole-2,3-dione, IST) has been used in analytical chemistry as a reagent for proline or tryptophan.1,2 In 1988, IST was discovered in human and rat urine by Glover et al.3 The increased IST concentration indicated the condition of neurological patients including stress, anxiety and epilepsy.4–6 Although the metabolic pathway of IST has not been fully clarified, speculations of IST biosynthesis are reported to be linked to the peroxidation of tryptophan7 and the effectiveness of the intestinal flora.8,9 A number of procedures have already been described for the measurement of IST in biological samples such as gas chromatography-mass fragmentometry (GC-MS)3,10 and HPLC with a spectrometric detector.11,12 GC-MS is useful in determining the existence of interesting substances; however, the need to maintain purity makes it unsuitable for routine work. Although HPLC is useful in analyzing a biological sample, its spectrophotometric detection is liable to lack sensitivity and/or selectivity. It requires a tedious pretreatment. For example, in the assay of Manabe et al.,11 the urine sample was treated by a liquid–liquid and solid-phase extraction, followed by preparative HPLC. As reported here, HPLC based on postcolumn photoirradiation was established with a Capcell Pak C1 column and with a mobile phase containing tetrahydrofuran and hydrogen peroxide. This method allowed easy pretreatment by dilution or deproteinizing and efficient measurements of IST in human urine and serum.Experimental
Chemicals and pooled serum
IST was obtained from Sigma (St. Louis, MO, USA) and was further purified by recrystallization from benzene. The standard IST sample was diluted using 0.1 mol l−1 hydrochloric acid and stored at −20 °C. All other chemicals were purchased from Wako (Osaka, Japan). Tetrahydrofuran (THF) was used as a solution free from stabilizer and was stored at 4 °C until the mobile phase was prepared. A freeze dried pooled serum (Consera®) was obtained from Nissui Seiyaku (Tokyo, Japan).Chromatographic system
The chromatographic system consisted of a high pressure pump (Model SSP DM-3U, Sanuki Kogyo, Tokyo, Japan), a valve injector (Model E1E005, Shimamura, Tokyo, Japan) fitted with a 200 µl loop, an analytical column, a photoreactor (Model S-3900, Soma Optics, Tokyo, Japan), a fluorescence monitor (Model RF-530, Shimadzu, Kyoto, Japan) with a 12 µl flow cell, and a Shimadzu Chromatopac C-R3A (Shimadzu, Kyoto, Japan) recorder–integrator. In this system the analytical column (250 × 4.6 mm id) was packed with Capcell Pak C1 (particle size 5 µm, Type SG-120, Shiseido, Tokyo, Japan). The S-3900 photoreactor consisted of a stainless steel photoreflector, an electric fan and two tubes of GL-4 germicidal light (4 W electric power; Nippon Denki, Tokyo, Japan). The photoirradiation was carried out in ETFE (a copolymer of ethylene and tetrafluoroethylene: Tefzel®, GL Sciences, Tokyo, Japan) tubing (0.25 mm id × 1.59 mm od, 4 m long) which was wound around each germicidal light tube. Following the photoirradiation coil, the back-pressure tubing was composed of PEEK (polyether ether ketone, GL Sciences, Tokyo, Japan) tubing (0.13 mm id × 0.5 m long) and PTFE tubing (0.25 mm id × 2 m long) was connected. The fluorescence of the effluent solution was measured with excitation and emission wavelengths of 302 nm and 418 nm, respectively. Chromatography was performed at ambient temperature.Separation conditions
The mobile phase, which consisted of 70 mmol l−1 phosphate buffer (pH 7.2) containing 5 mmol l−1 hydrogen peroxide and 15% (v/v) tetrahydrofuran, was delivered at a flow rate of 0.56 ml min−1.Analytical procedure for human urine and serum
IST was diluted with hydrochloric acid to maintain its stability and to solubilize any sediment in the biological sample. The human urine of healthy subjects (ordinary diet) was collected over a 24 h period in the vessel containing 10 ml of hydrochloric acid per liter of urine. The urine samples were stored at −20 °C until analysis.One milliliter of 5 mol l−1 hydrochloric acid was added to the urine (1 ml) and then heated for 10 min in boiling water. After the urine sample was cooled in an ice–water bath, the sample was diluted 50-fold with distilled water. An aliquot (20 µl) of the sample was injected into the previously described system. For the serum samples, 200 µl of 5 mol l−1 hydrochloric acid was added to the serum (200 µl) and then heated for 10 min in boiling water followed by cooling in an ice–water bath. The mixture was centrifuged at 9600g for 1 min.
One hundred microliters of 1.5 mol l−1 perchloric acid were added to 200 µl of the heated serum in a polypropylene tube (1.5 ml). The mixture was then vortex-mixed. The entire mixture was added to 100 µl of 1.5 mol l−1 potassium chloride. After being vortex-mixed and centrifuged for 1 min, a 50 µl portion of the supernatant fluid was injected into the chromatograph. The recovery test was carried out to observe the influence of the biological component on the fluorophore. The spiked urine or serum was prepared as follows. One milliliter of IST standard solution (854 ng ml−1) was added to 0.5 ml of the heated urine in the glass measuring flask which was filled to 25 ml with distilled water. Then, 1.5 mol l−1 perchloric acid solution was added to the serum containing the IST of 49.7 ng ml−1 concentration and deproteinized by the method described above. The peak heights obtained for the spiked sample were compared with those of the standard sample.
Results and discussion
Fluorescence reaction upon irradiation
Fig. 1 shows the excitation (a) and emission spectra (b) for the IST obtained using the LC system. The excitation (λex) and emission (λem) maxima were at 302, and 392 and 418 nm, respectively. IST is converted to anthranilic acid (2-aminobenzoic acid) in the presence of sodium hydroxide and hydrogen peroxide.13 In order to produce a fluorophore, the products were developed with the system described except for photoirradiation and two peaks were confirmed. The retention time of main and minor products were 7.6 min (λex 312 nm, λem 388 nm) and 9.0 min (λex 302 nm, λem 418 nm), respectively. The characteristics of the main product agreed with those of anthranilic acid. Although the minor product is still unknown, the spectrum of IST is obtained with two fluorophores.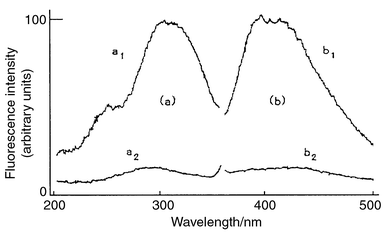 | ||
| Fig. 1 Fluorescence excitation (a) and emission (b) spectra of the HPLC eluate of IST. a1 and b1, 12.8 µg ml−1; a2 and b2, sample blank for a1 and b1, respectively. | ||
Optimization of postcolumn photoirradiation
The phosphate buffer for the maximum fluorescence intensity in the mobile phase was observed at pH 6.8–8.0. pH 7.2 was adopted based on the peak shape described in Optimization of chromatographic conditions (below) and the stability of the mobile phase. Table 1 shows the effects of organic solvents on the fluorescence intensity. The usual organic modifiers, such as methanol and acetonitrile, did not provide adequate sensitivity. If THF was added after the photoirradiation, it was not effective. The best range of concentrations in the mobile phase for the photoirradiation reaction was 15–30% v/v. For the separation of IST in biological samples, the concentration of 15% v/v THF was adopted. The optimum concentration of hydrogen peroxide were found to be 3–8 mmol l−1 for the 4.0 m long ETFE tubing while 5 mmol l−1 was adopted for additional experiments. The addition of hydrogen peroxide enhanced the fluorescence intensity by about 20%.| Organic modifier (20% v/v) | Relative fluorescence intensity (%) |
|---|---|
| None | 2.6 |
| Methanol | 6.5 |
| Ethanol | 8.8 |
| Propan-1-ol | 6.0 |
| Propan-2-ol | 5.9 |
| Acetonitrile | 12.9 |
| Tetrahydrofuran | 100 |
Optimization of chromatographic conditions
When the mobile phase which contained the 15% (v/v) tetrahydrofuran in 70 mmol l−1 phosphate buffer (pH 7.2) was used, the IST retention times with Capcell Pak C1, C8, C18 and CN (size 150 × 4.6 mm id) were 10, 8.6, 8.6 and 8.8 min, respectively. The Capcell Pak C1 column provided the best separation of IST, anthranilic acid (degradation product of IST) and coexisting components in the urine sample. The size of the adapted C1 column was 250 × 4.6 mm id to ensure separation with injection of a large volume of samples and with the serum sample. The IST retention time was not significantly influenced by the pH of the phosphate buffer in the eluent. The pH affected the peak shape of IST. Fig. 2 shows the effect of the pH of the phosphate buffer on the peak shapes. The range of pH 4.5–6.5 gave a broad and fronting peak shape, which almost became symmetrical at pH 7.2. The hydrolysis of IST has been reported by Casey et al.,14i.e., above pH 6 IST undergoes cleavage of the amide bond and forms an open ring structure, while at pH 4 there is an approximately equal ratio of the closed- and opened-ring forms.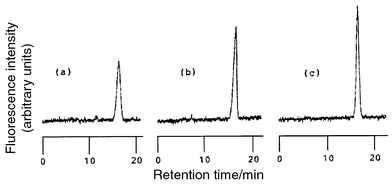 | ||
| Fig. 2 Effect of pH on the peak shapes of isatin. (a) pH 5.9, (b) pH 6.5 and (c) pH 7.2 in 70 mmol l−1 phosphate buffer. | ||
The fronting peak of IST appears during the acidified mobile phase.12 As the IST of the open-ring form showed a symmetrical peak, the phosphate buffer in the eluent was kept at pH 7.2. In addition, several compounds such as tryptophan, melatonin, kynurenine, kynurenic acid, anthranilic acid (retention time 7.6 min), nicotinic acid, tyrosine, phenylalanine, histidine, pyridoxine and thiamine did not interfere with the analysis although they were eluted faster than the IST peak.
Calibration and precision
The measured relative fluorescence intensity was linear with the amount of IST over the range of 0.16–10.7 ng (1.1–72.7 pmol). The correlation coefficient was 0.998, the slope was 26.8 and the intercept was −0.8 using a least squares regression analysis. The parameters y and x are the relative fluorescence intensity in % and the amount of IST in ng, respectively. The relative standard deviation of the standard IST was 1.9% at 0.62 ng (n = 8) and 1.2% at 4.0 ng (n = 8) . The detection limit (signal to noise = 3) was determined to be 0.11 ng (0.75 pmol).Selectivity and recovery
Fig. 3 shows chromatograms of authentic IST and urine samples. The retention time of IST was 16 min. The IST peak was observed in chromatograms (a), (b) and (c) at the corresponding retention times, whereas a chromatogram (d) of the unirradiated sample showed no peak at that position. Furthermore, the spectrum of the peak of interest was similar to that of IST (Fig. 4). These comparisons of the chromatograms demonstrate the specificity of the present method. The recovery (±s) of IST added to urine (854 ng per 25 ml of urine) was 96.3 ± 4.7% (n = 7). In the urine of six normal subjects, the range and mean ±s of urinary IST was 184–1513 µg l−1 and 735.4 ± 491.5 µg l−1, respectively. This value in human urine was similar to the 708.4 ± 766.9 µg l−1 reported by Manabe et al.11Fig. 5 compares the chromatograms of irradiated and non-irradiated samples in deproteinized serum. | ||
| Fig. 3 Chromatograms obtained with a standard and the urine samples. Irradiation with UV light: (a) standard of IST (0.68 ng); (b) urine sample; (c) urine sample spiked with IST;. (d) the same sample as in (b), but without irradiation. Analytical procedure and conditions as described in the Experimental section. Injection volume was 20 µl. Applied concentration of IST was 34.1 ng ml−1 of diluted urine. | ||
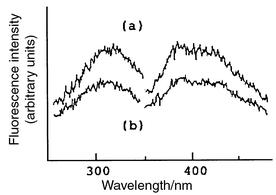 | ||
| Fig. 4 Fluorescence excitation and emission spectra of the HPLC eluate of (a) standard peak (20.8 ng ml−1) and (b) component of IST peak in urine sample. Spectra determined by Shimadzu Spectrofluorometer RF-5000. | ||
 | ||
| Fig. 5 Chromatograms obtained with a standard and the serum samples. Irradiation with UV light: (a) standard of IST (0.62 ng); (b) serum sample; (c) serum sample spiked with IST; (d) the same sample as in (b), but without irradiation. Analytical procedure and conditions were as described in the Experimental section. Injection volume was 50 µl. Applied amount of IST was 49.7 ng in deproteinized serum. | ||
The assay of the serum was sufficient using 200 µl and is possible at 1/25 the volume compared with UV detection. The spectra of the IST peak was similar to that shown in Fig. 4. The recovery (±s) of IST added to serum (49.7 ng ml−1 in perchloric acid) was 94.3 ± 4.4% (n = 6). The mean ±s of serum IST was 54.7 ± 4.2 ng ml−1 (n = 6). For reference, the value in human plasma was 134.2 ± 120.6 ng ml−1 as reported by Manabe et al.11 The variation in the IST level is presumed to correspond to the difference in dietary tryptophan content and might be influenced by intestinal flora.
This method is both sensitive and sufficiently specific to estimate IST in human urine and serum and should be useful in biochemical and clinical studies.
Acknowledgment
The authors acknowledge the lending of the photoreactor (S-3900) from Soma Optics.References
- F. N. Boctor, Anal. Biochem., 1971, 43, 66 CrossRef CAS.
- C. I. Trigoso, N. Ibanez and J. C. Stockert, J. Histochem. Cytochem., 1993, 41, 1557 Search PubMed.
- V. Glover, J. M. Halket, P. T. Watkins, A. Clow, B.L. Goodwin and M. Sandler, J. Neurochem., 1988, 51, 656 CAS.
- V. Glover, S. K. Bhattacharya and M. Sandler, Indian J. Exp. Biol., 1991, 29, 1 Search PubMed.
- D. Hota and S. B. Acharya, Indian J. Exp. Biol., 1994, 32, 710 Search PubMed.
- N. Hamaue, Yakugaku Zasshi, 2000, 120, 352 Search PubMed.
- S. Ghosal, S. K. Bhattacharya, A.V. Muruganandam and K.S. Satyan, Biogenic Amines, 1997, 13, 91 Search PubMed.
- A. H. Jackson, R. T. Jenkins, M. Grinstein, A.-M. Ferramola de Sancovich and H. A. Sancovich, Clin. Chim. Acta, 1988, 172, 245 CrossRef CAS.
- M. Sandler, A. Przyborowska, J. Halket, P. Watkins, V. Glover and M. E. Coates, J. Neurochem., 1991, 57, 1074 CAS.
- J. M. Halket, P. J. Watkins, A. Przyborowska, B. L. Goodwin, A. Clow, V. Glover and M. Sandler, J. Chromatogr., 1991, 562, 279 CrossRef CAS.
- S. Manabe, Q. Gao, J. Yuan, T. Takahashi and A. Ueki, J. Chromatogr. B, 1997, 691, 197 CrossRef CAS.
- N. Hamaue, N. Yamazaki, M. Minami, T. Endo, M. Hirahuji, Y. Monma and H. Togashi, Gen. Pharmac., 1998, 30, 387 Search PubMed.
- W. J. Houlihan, in The Chemistry of Heterocyclic Compounds, Indoles, eds. A. Weissberger and E. C. Taylor, Wiley, New York, 1972, pt. 1, ch. 1, pp. 157–158. Search PubMed.
- L. A. Casey, R. Galt and M. I. Page, J. Chem. Soc., Perkin. Trans., 1993, 2, 23 Search PubMed.
| This journal is © The Royal Society of Chemistry 2001 |