
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot organocatalyzed synthesis of tricyclic indolizines†
Lucas A.
Zeoly
 a,
Lais V.
Acconcia
a,
Manoel T.
Rodrigues
Jr.
a,
Hugo
Santos
a,
Rodrigo A.
Cormanich
a,
Juan C.
Paniagua
bc,
Albert
Moyano
*d and
Fernando
Coelho
*a
a,
Lais V.
Acconcia
a,
Manoel T.
Rodrigues
Jr.
a,
Hugo
Santos
a,
Rodrigo A.
Cormanich
a,
Juan C.
Paniagua
bc,
Albert
Moyano
*d and
Fernando
Coelho
*a
aLaboratório de Síntese de Produtos Naturais e Fármacos, Department of Organic Chemistry, Institute of Chemistry, University of Campinas, PO Box 6154-13083-970, Campinas, SP, Brazil. E-mail: facoelho@unicamp.br
bDepartament de Ciència de Materials i Química Física, Universitat de Barcelona, Facultat de Química, C. Martí i Franquès 1-11-08028, Barcelona, Spain
cInstitut de Química Teòrica i Computacional, Universitat de Barcelona, Spain
dDepartament de Química Inorgànica i Orgànica, Universitat de Barcelona, Facultat de Química, C. Martí i Franquès 1-11-08028, Barcelona, Spain
First published on 14th April 2023
Abstract
Indolizines and their saturated derivatives are important structural motifs present in several biologically active compounds of both natural and synthetic origin. We describe herein a one-pot approach for the synthesis of tricyclic indolizines catalyzed by a bicyclic imidazole-alcohol. The protocol is based on an aqueous Morita–Baylis–Hillman reaction between pyridine-2-carboxaldehydes and six- or seven-membered cyclic enones, followed by sequential intramolecular cyclization and dehydration. So, in a single operational step two new bonds (C–C and C–N) are formed in an organocatalyzed process that takes place in simple conditions (stirring in water at 60 °C for 12 h) and with great atom economy (water as the sole byproduct), affording the purified compounds in yields ranging from 19 to 70%. The facility of the cyclization strongly depends on the size of the cycloalkenone ring: while MBH adducts derived from six-, seven- or eight-membered cycloenones are readily transformed into the corresponding indolizines, cyclopentenone-derived MBH adducts do not cyclize. A competition experiment revealed that cycloheptenone-derived MBH adducts cyclize faster than cyclohexenone-derived adducts. Model DFT calculations have been performed to rationalize these reactivity trends.
Introduction
Nitrogenated heterocycles play an important role in several segments of the chemical and pharmaceutical industry.1 Indolizines are unsaturated nitrogen heterocycles resulting from the fusion of two heterocyclic nuclei: a pyridine and a pyrrole, in which the nitrogen atom is shared by both rings. This structural motif is present in biologically active products of both natural and synthetic origin,2 as well as in materials with other applications. Although the occurrence of the indolizine nucleus in natural products is rather scarce, their hydrogenated derivatives (either tetrahydroindolizines or indolizidines) are present in a variety of biologically important natural products, the best known of them being the so-called indolizidine alkaloids.3 The structural resemblance of indolizines with indoles has stimulated the incorporation of this heterocycle in the design of synthetic compounds with different biological activities, such as, anti-cancer drugs,4 anti-microbials,5 anti-inflammatory compounds,6 phosphatase inhibitors,7 and drugs to treat schizophrenia,8 among others.9 Moreover, several polycyclic indolizines are fluorescent, with applications as biological probes10 and in electronic devices11 (Fig. 1).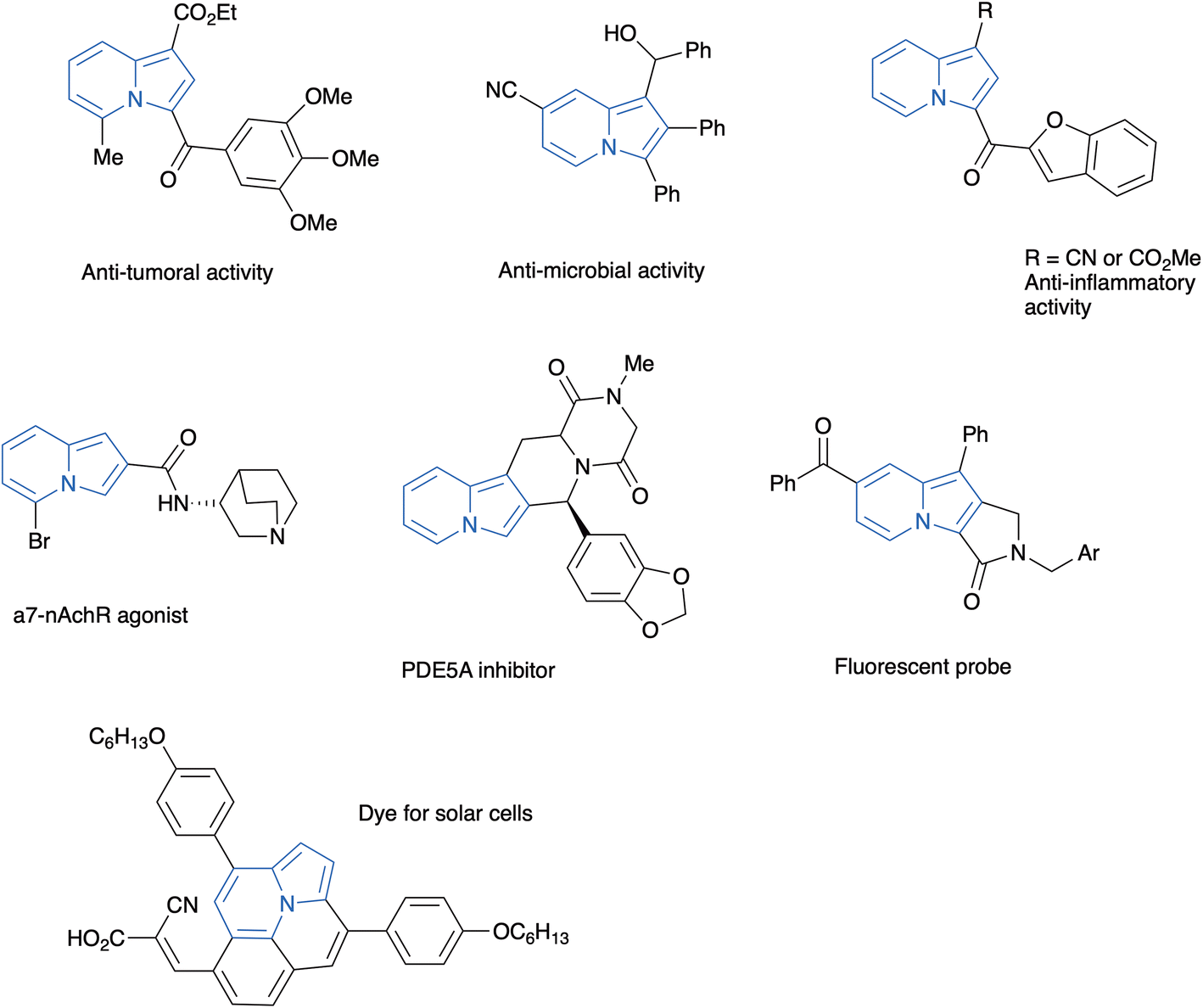 | ||
| Fig. 1 Some representative examples of biologically active and of fluorescent indolizines. | ||
Classically, indolizines have been prepared through Scholtz12 or Chichibabin reactions.13 However, the biological effects associated to the indolizine core and the interest for their photophysical properties have stimulated the development of a plethora of approaches to obtain this heterocyclic scaffold.8,14,15 Recently, due to the same reasons, different methods to prepare polycyclic indolizines have been developed.16
The Morita–Baylis–Hillman (MBH) reaction17 is a sustainable chemical transformation that provides access to small, functionalized molecules, and which has proven to be an efficient platform for the synthesis of the indolizine framework.18 Polycyclic indolizines have also been synthesized by exploring the synthetic versatility of MBH adducts. However, in most instances it is necessary to use an acetylated18a,b,f,h,j,k or a brominated derivative (Scheme 1a).18f,g In the few cases where the adducts are used directly, they must usually be activated in situ by means of a Lewis acid18c,i or by using special reaction conditions, such as microwave irradiation (Scheme 1b).18d On the other hand, the MBH reaction has very seldom been used in a one-step, two-component synthesis of indolizines in a purely organocatalyzed process.19 Batra and co-workers18f reported that without previous conversion to a bromide, the MBH adducts of cycloenones with N-substituted 1-formyl-β-carbolines cyclized very slowly (4 days in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 THF–H2O at rt) to the corresponding indolizino-indole derivatives. A single example was reported, with an isolated yield of 58%. We wish to disclose presently that the bicyclic imidazolyl alcohol (BIA) 1, an extremely convenient catalyst for the aqueous MBH reactions of conjugated cycloalkenones,20,21 can be advantageously put to use in the direct organocatalyzed synthesis of a variety of tricyclic indolizines via a MBH/aza-Michael/dehydration cascade (Scheme 1c).
:1 THF–H2O at rt) to the corresponding indolizino-indole derivatives. A single example was reported, with an isolated yield of 58%. We wish to disclose presently that the bicyclic imidazolyl alcohol (BIA) 1, an extremely convenient catalyst for the aqueous MBH reactions of conjugated cycloalkenones,20,21 can be advantageously put to use in the direct organocatalyzed synthesis of a variety of tricyclic indolizines via a MBH/aza-Michael/dehydration cascade (Scheme 1c).
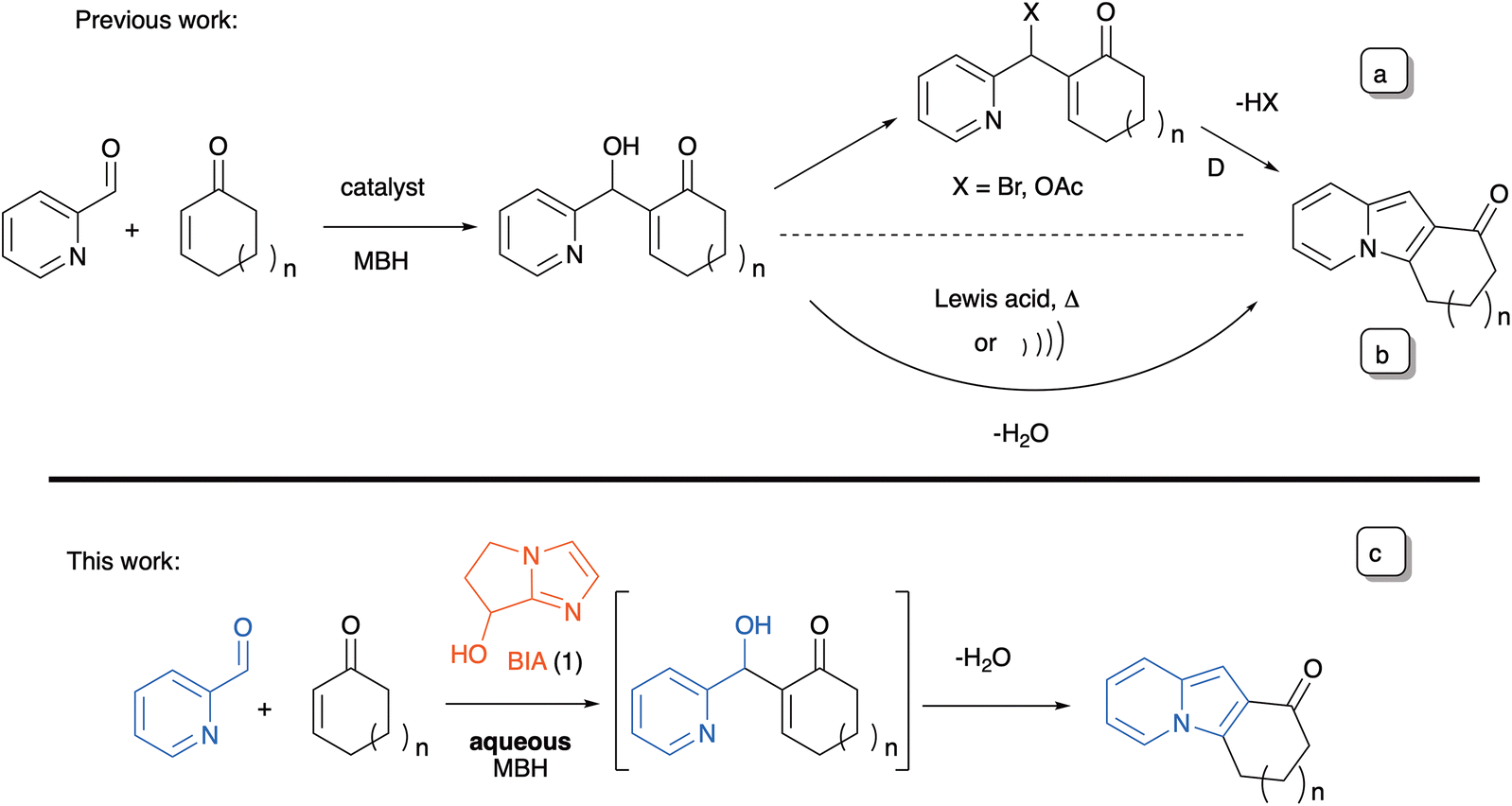 | ||
| Scheme 1 Research background. | ||
Results and discussion
Our research began with the observation that the BIA-catalyzed aqueous MBH reaction between 2-pyridinecarbaldehyde (2a) and 2-cyclohexenone (3a) consistently afforded mixtures of the expected MBH adduct 4aa18k,22 and of the tricyclic indolizine 5aa, even at ambient temperature and with short reaction times (Table 1, entry 1). It is worth noting here that the reaction of 2a and 3a in the conditions described by Batra (0.5 equiv. DMAP, 1:1 THF–H2O, rt)18f required 4 days for completion and afforded 5aa in 40% yield.18b
|
|
|||||
|---|---|---|---|---|---|
| Entry | Lewis base (equiv./solvent) | Ratio 4aa/5aaa | Yieldb (%) | ||
| 3 h | 24 h | 48 h | |||
| a Ratio between the peaks corresponding to MBH adduct (4aa) and to indolizine (5aa), measured by analysis of relative areas (GC-MS). b Yield of 5aa after 48 h, calculated by 1H-NMR, using 1,2,4,5-tetrachloro-3-nitrobenzene as internal standard. c SDS = sodium dodecyl sulfate. d Conversion of 2a was already complete at this point. e The conversion was too low to calculate the area ratio. f Total conversion of 2a was achieved at this point. g Not determined. | |||||
| 1 | BIA (0.65/H2O-10 mol% SDSc) | 58:42d |
22:78 |
6:94 |
68 |
| 2 | 3-Quinuclidinol (0.65/H2O) | 74:26 |
26:74 |
8:92 |
68 |
| 3 | Triethylamine (1.0/H2O) | 58:42 |
36:64 |
11:89 |
51 |
| 4 | Imidazole (1.0/aq. sat. NaHCO3) | —e | 51:49 |
49:51f |
42 |
| 5 | DBU (1.0/H2O) | —e | —e | —e | —g |
| 6 | TMPDA (1.0/H2O) | —e | 94:6 |
94:6 |
—g |
| 7 | NaOMe (1.0/MeOH) | —e | —e | —e | —g |
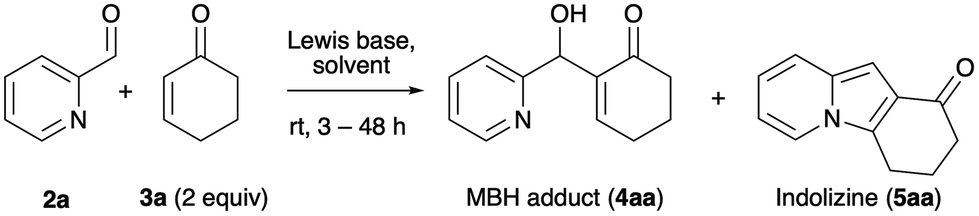
Among the catalysts classically employed in MBH reactions, only BIA (Table 1, entry 1), 3-quinuclidinol (entry 2), triethylamine (entry 3), and imidazole (entry 4) were able to form indolizine 5aa in good yield after 48 h at rt. Tetramethyl-1,3-propanediamine (TMPDA, entry 6) gave almost exclusively the MBH adduct, although in poor conversion. Both DBU (entry 5) and NaOMe in MeOH (entry 7) were not catalytically active at rt. BIA (entry 1) and 3-quinuclidinol (entry 1) exhibited the best ratios of conversion and yield. In the case of BIA, essentially complete conversion of the aldehyde 2a was reached in less than 3 h. Interestingly, both catalysts have a hydroxyl group in the proximity of the nucleophilic nitrogen atom, which has been shown to play a relevant role in the catalysis of the MBH reaction.23 This observation is also in accordance with the increase commonly observed in the MBH reaction rate when hydroxylic solvents are employed.24
We chose the conditions of entry 1 (0.65 equiv. BIA, 10% mol of sodium dodecyl sulfate, SDS in water) as the starting point from which to optimize the yield of the indolizine product. Our choice was based on the observation that BIA gave a better indolizine/MBH adduct ratio than quinuclidinol. Additionally, this bifunctional Lewis base is easily prepared in large scale using cheap starting materials.21,25 Thus, starting from these standard conditions (which had been previously optimized for the aqueous MBH additions of aryl aldehydes to cycloalkenones)20 we evaluated different solvents, temperatures, and equivalents of surfactant (SDS). The reactions were run both in protic and in aprotic solvents, such as iPrOH, AcOH, MTBE, CHCl3, HMPA and fluorobenzene. We were not able to detect indolizine formation in any of these. Note that attempts to promote the reaction with sodium methoxide in methanol (Table 1, entry 7) were also unsuccessful. So, we kept water as the solvent and evaluated the effect of the reaction temperature in the conversion. We ran reactions at 40 °C (67% of yield of 5aa after 24 h), 60 °C (70% yield after 12 h) and 100 °C (52% after 12 h). Based on these results, 60 °C was established as the best temperature. The amount (20 mol% or 30 mol%) and type (CTAB, Triton-X100) of surfactant were also changed, however no modification in yield was observed, so that we kept the concentration of SDS at 10 mol%.
With the optimal condition in hands, we started the evaluation of the reaction scope, using a set of 2-cycloalkenones of different ring size and substitution degree (Fig. 2). 2-Cyclohexenone (3a), 2-cyclopentenone (3b), and 2-cycloheptenone (3c) were commercially available; the remaining enones (5,5-dimethyl-cyclohexenone (3d), 5-phenyl-cyclohexenone (3e), 5-piperonyl-cyclohexenone (3f), and 2-cyclooctenone (3g)) were prepared according to previously described protocols.26,27
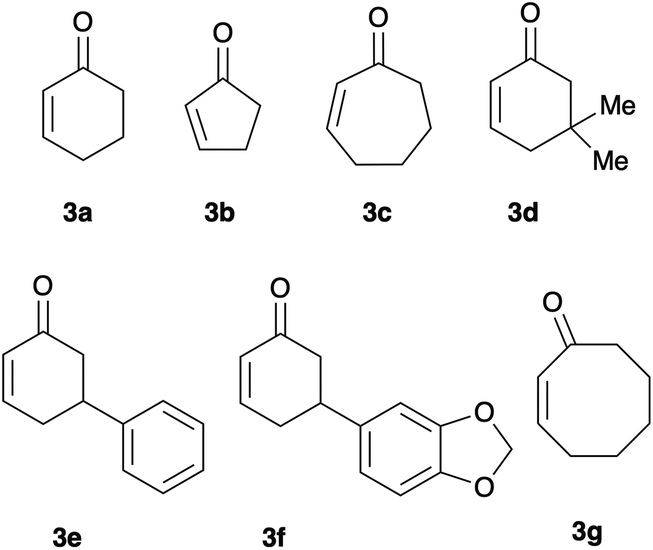 | ||
| Fig. 2 Cycloalkenones 3a–g chosen to evaluate the scope of the reaction. | ||
A set of commercial 2-pyridinecarboxaldehydes with different substitution patterns (2a–2i; Scheme 2) were treated with 2 mol equiv. of cycloalkenones in the presence of BIA (1; 0.65 mol equiv.) in water and SDS (0.1 mol equiv.), at 60 °C during 12 h.
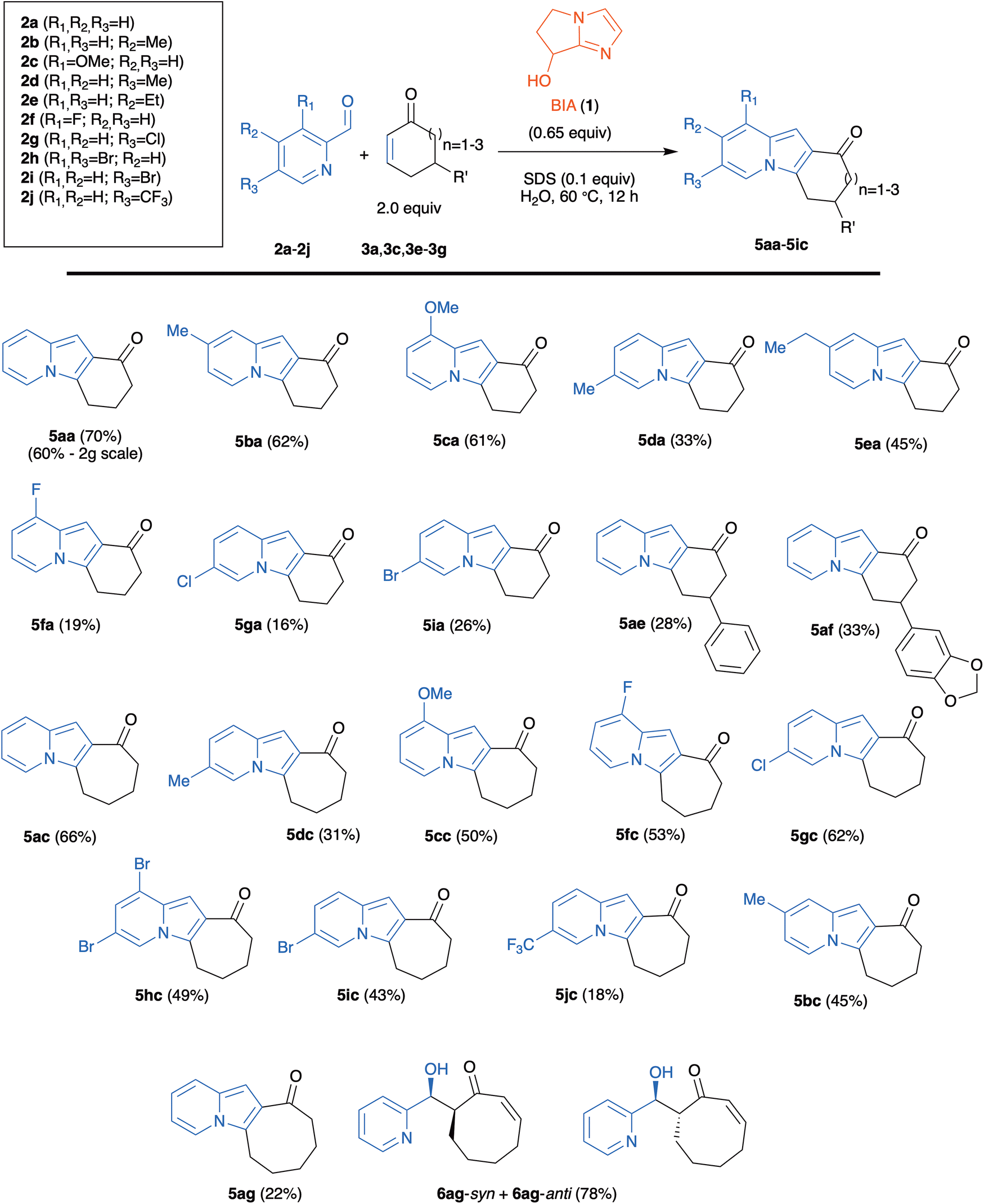 | ||
| Scheme 2 One-pot organocatalyzed synthesis of tricyclic indolizines. % Yields refer to isolated products after chromatographic purification. | ||
The reaction worked nicely with cyclohexenones 3a, 3e, 3f and with cycloheptenone 3c to afford the corresponding tricyclic indolizines 5 in one step and in moderate to good yields (Scheme 2). The yields with cycloheptenone 3c (ranging from 31% up to 66%) were in several instances higher than those obtained with cyclohexenones (ranging from 16% to up 70%), especially for reactions with halogen-substituted 2-pyridinecarboxaldehydes (compare 5fa with 5fc, 5ga with 5gc, and 5ia with 5ic). The introduction of a substituent in the 5-position of 3a gave rise to diminished yields (compare 5ae and 5af with 5aa). The reactions with 5,5-dimethyl-2-cyclohexenone 3d failed, as we did not detect the initial formation of the MBH adducts (probably due to the steric hindrance to the initial Michael addition of the nucleophilic catalyst).23 A single example with 2-cyclooctenone 3g was also examined. The low yield (22%) of the 2-cyclooctenone derivative 5ag was due to the low MBH reactivity of 3g, that led to the competitive formation (in 78% global yield) of aldol adducts 6ag-syn and 6ag-anti (which obviously cannot cyclize to the indolizine). It is worth noting therefore that the initially formed MBH adduct was converted quantitatively to the tricyclic indolizine 5ag.
4-Methyl-substituted aldehyde 2b gave, both for reactions with 2-cyclohexenone 3a and with 2-cycloheptenone 3c, better yields than the 5-methyl-substituted isomer 2d (compare 5ba with 5da, and 5bc with 5dc in Scheme 2). In the case of aldehyde 2j, with a strongly electron-withdrawing trifluoromethyl substituent at C-5, the reaction with 3c gave the tricyclic indolizine 5jc in very low yield (18%), while the reaction with 3a exclusively afforded the MBH adduct 4ja in 75% yield (Scheme 3).
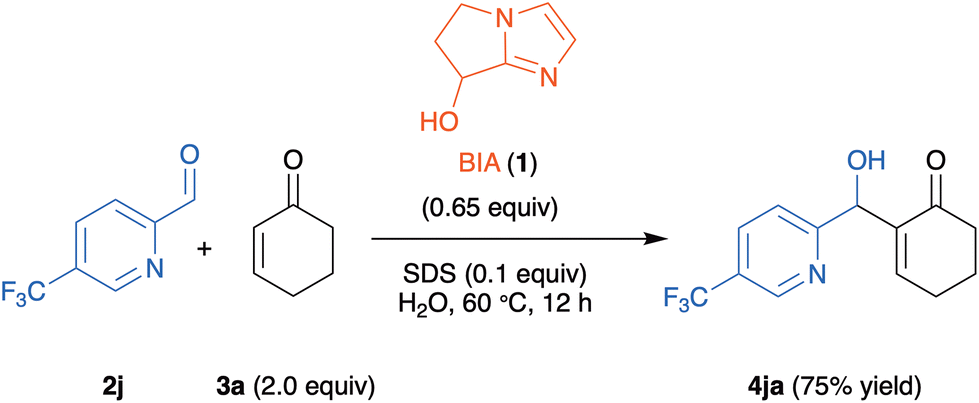 | ||
| Scheme 3 MBH adduct 4ja obtained in the reaction of 5-trifluoromethyl-2-pyridinecarboxaldehyde 2j with 2-cyclohexenone 3a. | ||
Even if in some instances (particularly in the reaction of 2-pyridinecarboxaldehydes bearing electron-attracting substituents with 2-cyclohexenones) yields are lower than 40%, this operationally simple, one-step process meets a series of green chemistry requirements, such as the use of water as solvent, high atom economy, the use of a cheap organic compound as the catalyst, and the possibility of recycling the reagents used in excess and the Lewis base, if necessary. It is also worth noting that the reaction can be used for the gram-scale synthesis of indolizines, as exemplified by the preparation of 5aa (2 g) in 60% yield (see Scheme 2).
On the other hand, the cyclization to the indolizine did not take place when 2-cyclopentenone 3b was employed as the Michael acceptor, independently of the nature of the pyridine-2-carboxaldehyde partner. For these reactions, we could only isolate the corresponding MBH adducts 4 mixed in some instances with the aldol products 6. Three representative examples are shown in Scheme 4. We were unable to detect the formation of the corresponding indolizines 5. Attempts to circumvent this issue, either by heating the reactions or by increasing the catalyst loading, failed. This indicates that MBH adducts derived from 3b do not cyclize to the corresponding indolizines in aqueous solution, a behavior with strongly contrasts with that of the other enones with six, seven or eight-membered rings.
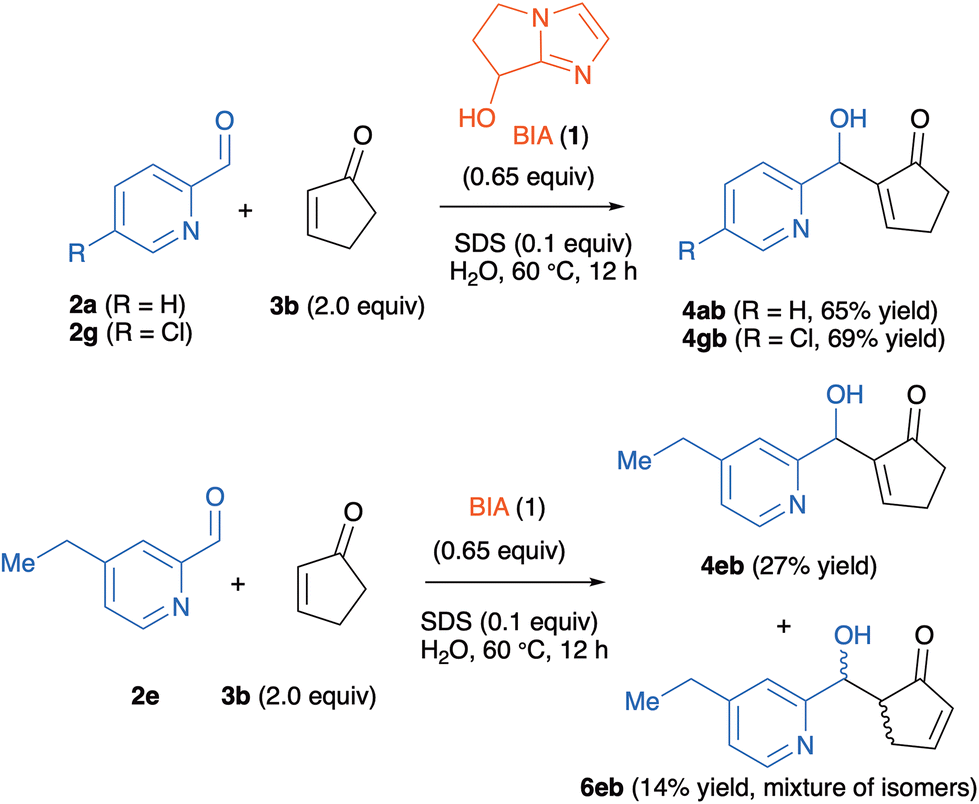 | ||
| Scheme 4 MBH and aldol adducts obtained from the reactions of 2-pyridinecarboxaldehydes 2a, 2e and 2g with 2-cyclopentenone 3b. | ||
Having established the scope of the reaction, we devoted our attention to the mechanism of this transformation. An important fact revealed by the data gathered in Table 1 is that the 4aa/5aa ratio is dependent on the nature of the promoter of the MBH reaction, raising the question of the eventual catalysis of the cyclization/dehydration step by the same compound. To ascertain this issue, we checked if the rate of the cyclization of pure 4aa (that could be obtained by stopping the BIA-catalyzed reaction after 1 h, followed by fast chromatographic purification of the product mixture in silica gel) in water was affected by the presence of the Lewis base catalyst. In fact, when a 0.1 M aqueous solution of MBH adduct 4aa was stirred at 25 °C, and the evolution of the system was monitored by 1H NMR spectroscopy, a similar conversion to 5aa (ca. 32% after 2 h) was observed with: (a) 0.65 equiv. of BIA, (b) 0.65 equiv. of 3-quinuclidinol, and (c) in the absence of a Lewis base. No further conversion of 4aa to 5aa took place in CDCl3 solution. This shows therefore that the role of the BIA catalyst 1 is restricted to promoting the MBH addition,28 and that the cyclization/dehydration of the MBH adducts in water is much faster than in other solvents (including THF/H2O mixtures).
Particularly in the case of electron-deficient 2-pyridinecarboxaldehydes, 2-cycloheptenone-derived MBH adducts appear to cyclize more efficiently than those obtained from 2-cyclohexenone. To collect more evidence about the reactivity of the cycloalkenones in the cyclization step, we decided to carry out a competition experiment. We selected for this purpose the reaction between 3-fluoro-2-pyridinecarboxaldehyde (2f) and a 1:1 mixture of 2-cyclohexenone 3a and 2-cycloheptenone 3c in water/DMSO (Scheme 5), for the following reasons: (a) the possibility of following the reaction by 19F NMR, (b) electron-poor aldehydes give good yields of MBH adducts, and (c) the clear difference in yields observed between 5fa and 5fc in Scheme 2 suggested that the rates of formation of both adducts would also be very different.
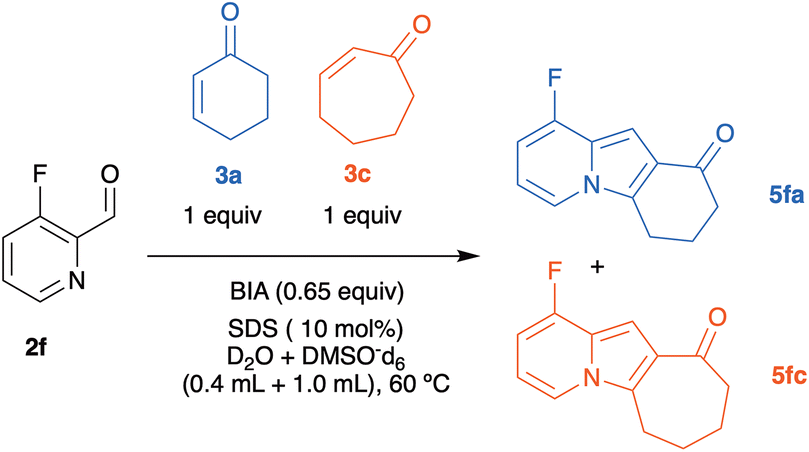 | ||
| Scheme 5 Competition experiment between 3a, 3c and the fluorinated pyridinecarboxaldehyde 2f. | ||
The reaction was monitored using a pseudo-2D experiment directly in the NMR tube. Both enones were added at the same time and the tube was then brought to the spectrometer with the probe temperature already set at 60 °C. The spectra were collected for 6 h with a 10-minute interval between each acquisition and the result obtained for the formation of indolizines and the consumption of 3-fluoro-2-pyridinecarboxaldehyde are shown below (Fig. 3). We opted for monitoring the reaction by 19F NMR, due to the high sensibility of this nucleus to small structural changes. Even though the fluorine atom is in the aromatic ring, the 19F chemical shift in the indolizine 5fa prepared from cyclohexenone (−125.5 ppm) is different from that of 5fc, arising from cycloheptenone (−125.0 ppm). Analysis of Fig. 3 shows that the concentration of the pyridinecarboxaldehyde 2f decreases continuously during the first 6 h of the reaction, and that the cycloheptenone-derived indolizine 5fc is formed faster than the cyclohexanone-derived indolizine 5fb (whose concentration does not change after 3 h from the beginning of the reaction). This shows that as we had anticipated in this case the rate of the cyclization step is therefore determining the overall rate of the process. Therefore, the reactivity order for the cyclization of the MBH adducts 4 to the indolizines 5 with respect the ring size of the enones is:
| cycloheptenone > cyclohexenone ≫ cyclopentenone |
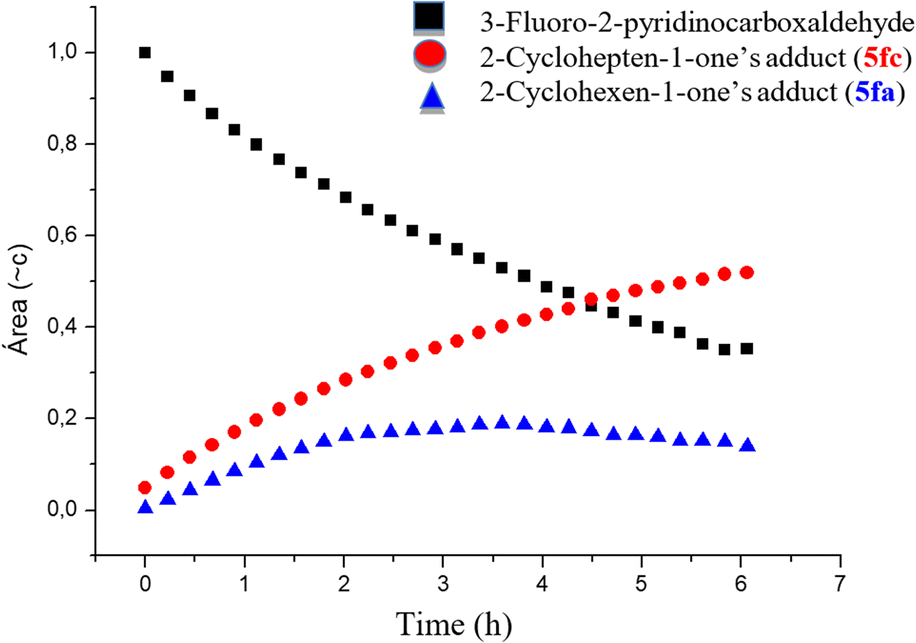 | ||
| Fig. 3 Evaluating the relative rate of cyclization reaction using six and seven-membered enones (graph area vs. time obtained from analysis of the 19F NMR spectra). | ||
On the other hand, the fact that the cyclooctenone-derived indolizine 5ag is formed in essentially quantitative yield from the corresponding MBH adduct 4ag (see data in Scheme 2) suggests that MBH adducts of cyclooctenone cyclize even faster than those of cycloheptenone.
In summary, experimental evidence suggests that the efficiency of the bifunctional catalyst 1 in promoting the direct conversion of 2-pyridinecarbaldehydes and cyclic enones into the indolizine derivatives comes from the fact that the cyclization of the initially formed MBH adducts takes place in aqueous solution without the need of previous activation of the hydroxyl group or of acid catalysis and is accelerated by heating. The rate of the cyclization does not depend appreciably on the nature of the pyridinecarbaldehyde component but varies strongly with the ring size of the cycloenones.
A plausible mechanistic proposal for the conversion of the MBH adducts 4 into the indolizines 5 is that this key transformation takes place in two stages: (a) intramolecular Michael attack of the pyridine nitrogen to the enone moiety, leading to a cyclized zwitterionic intermediate (I), and (b) dehydration of I to afford the final product (Scheme 6). It is worth noting here that step (a) is a 5-endo-trig cyclization, a disfavored process according to Baldwin's rules for ring closure.29 This step should therefore be highly endothermic, and its rate should be controlled both by the strain energy of the tricyclic intermediate I and by the polarity of the solvent. On the other hand, the dehydration step (b), although strongly exothermic due to the formation of water and of the resonance stabilization energy of the aromatic indolizinic ketone, should have a high activation energy for a concerted pathway, and is a likely candidate for catalytic assistance by a protic polar solvent. We propose that a water molecule, upon hydrogen-bonding to the hydroxyl group of I, provides a relatively low-energy pathway for the elimination of hydroxide anion, leading to the cationic intermediate II that can easily lose a proton to give the tricyclic indolizine.
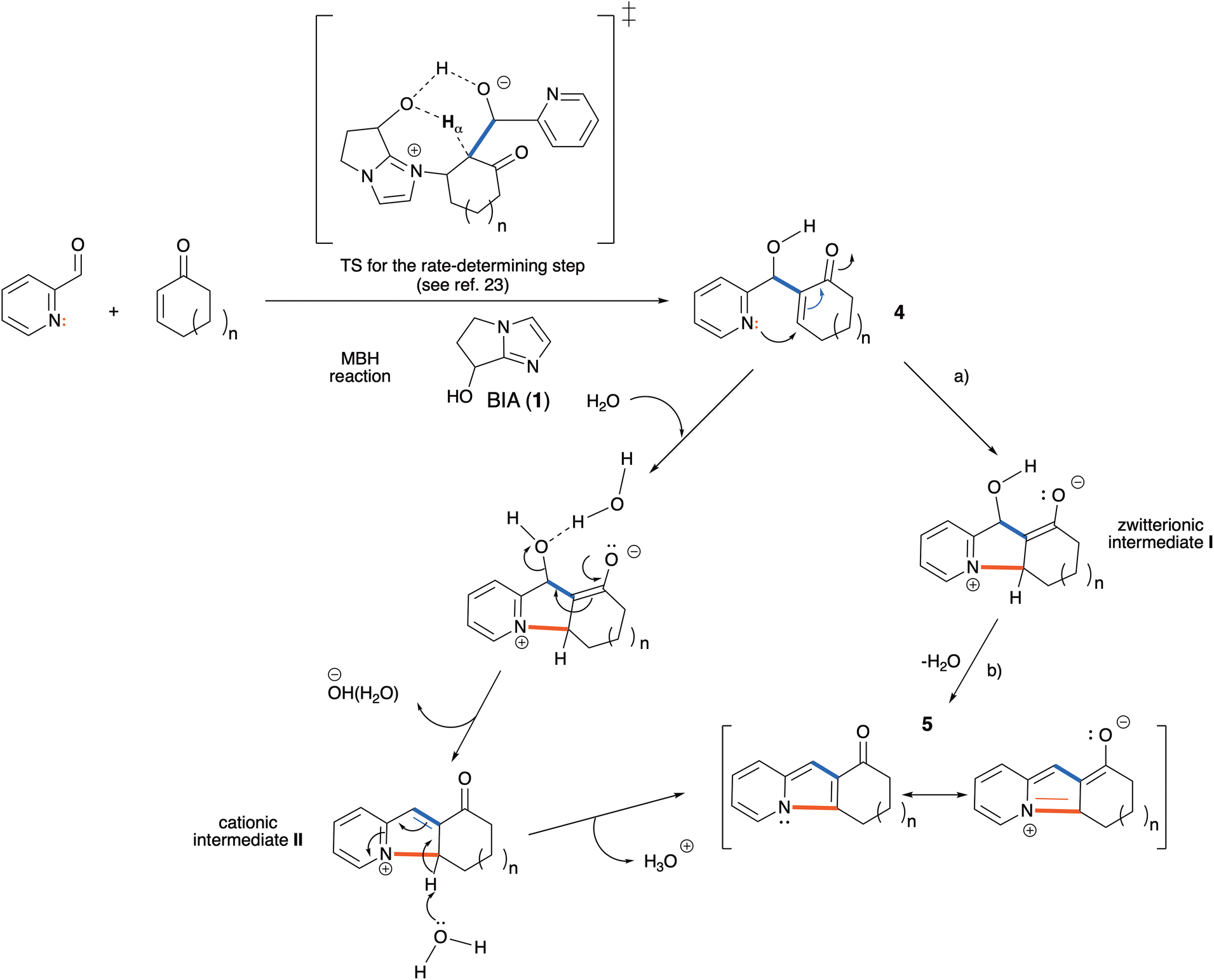 | ||
| Scheme 6 Plausible mechanism for the direct conversion of MBH adducts 4 into indolizines 5 in water. The two bonds formed in the overall process are showcased in blue (MBH step) and in red (aza-Michael/dehydration step). | ||
We have performed theoretical calculations that give initial support to this hypothesis, at the DFT level of theory and both in water solution (implicit solvent model) and in the gas phase (gp). We have located and characterized the transition states (TS) for step (a) (intramolecular aza-Michael addition) of the cyclization/dehydration with 2-cyclopentenone, 2-cyclohexenone and 2-cycloheptenone, respectively. The results are summarized below (Fig. 4). See the ESI† for additional details about the calculations.
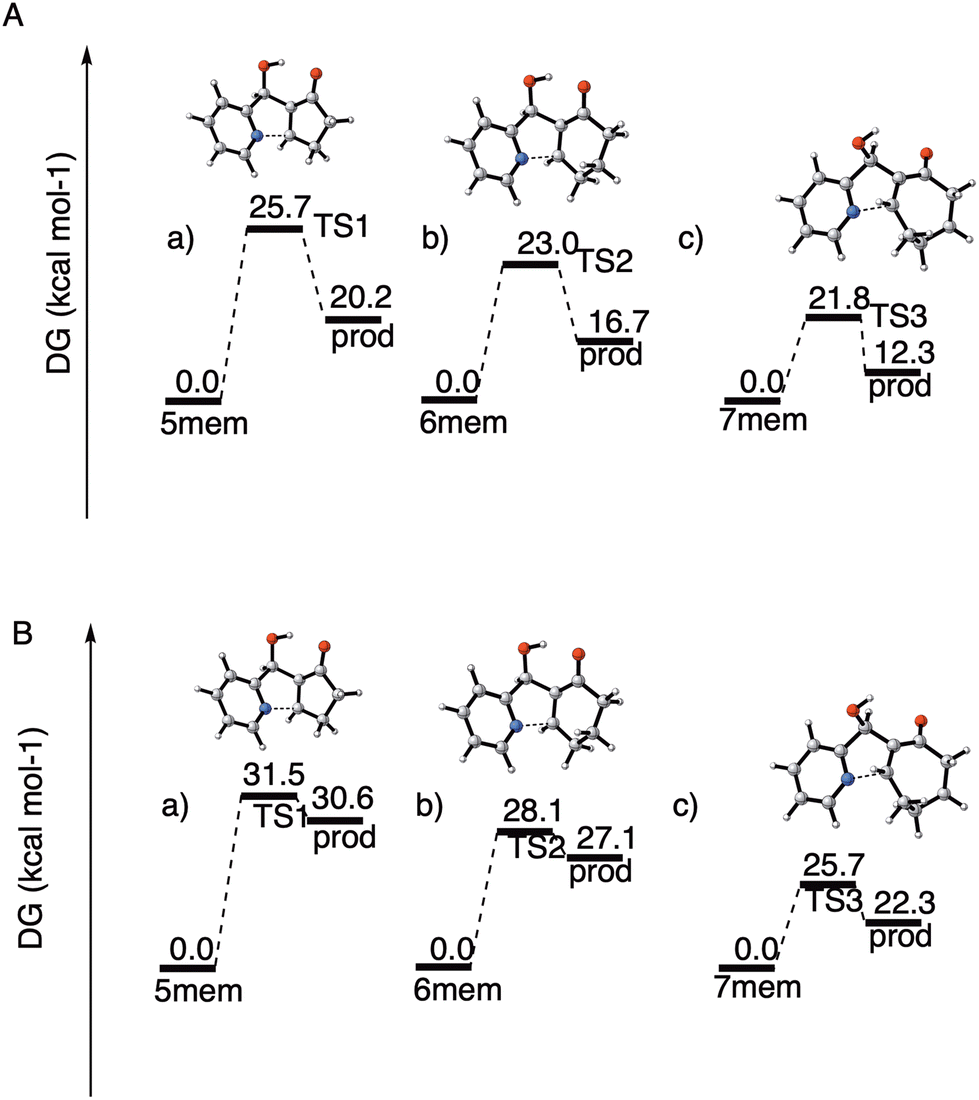 | ||
| Fig. 4 Calculated energy barriers for the ring closing step obtained at the ωB97xD/aug-cc-pVTZ theoretical level. (A) In water solution. (B) In the gas phase. | ||
In agreement with our hypothesis, the calculations show that in water solution step (a) is endoergic in all instances, and that the activation free energies can be clearly correlated with the difference in free energy between the MBH adduct and the zwitterionic intermediate I. As can be seen from Fig. 4A, the energy barrier for the formation of the 5-membered tricyclic indolizine is 2.7 kcal mol−1 higher than that for the formation of the 6-membered one, which in turn is 1.2 kcal mol−1 higher than that for the formation of the 7-membered one. According to these data, the cyclization of the five-membered MBH product 4ab is 1000 times slower than that of the seven-membered adduct 4ac, that in turn is only ten times faster than the cyclization of 4aa. These theoretical results corroborate our previous observations regarding the very poor reactivity of the 5-membered ring analogues, which hinders indolizine formation during the surveyed reaction time, and the higher reactivity of the 7-membered adduct when compared to the 6-membered adduct in the cyclization reaction.
To assess the effect of the aqueous solvent in the process, we also computed the activation free energies for the cyclization step in the gas phase, at the same theoretical level (Fig. 4B). When comparing the relative energies of the transition states, we found that the energy barriers associated to TS1, TS2 and TS3 were respectively 4.1, 5.9 and 4.9 kcal mol−1lower in water solution than in the gas phase. For the zwitterionic intermediates the effect of the solvent is even more pronounced, since in the gas phase their energies are systematically 10 kcal mol−1 higher than in water solution. The differential stabilization of the zwitterionic intermediates and of the corresponding transition states with respect to the starting MBH adducts upon solvation by a highly polar solvent is in accordance with the experimental observation that in non-aqueous media the direct cyclization of the MBH alcohols is very difficult and requires either the previous conversion of the hydroxyl moiety to a good leaving group or activation by means of a Lewis acid.18
At this point, we were also interested in evaluating the synthetic usefulness of these new polycyclic indolizines. Thus, indolizine 5aa was taken as a platform for some chemical transformations. Our aim was to explore the reactivity of the different parts of the structure of this heterocycle, i.e., the aromatic system, the benzylic position, the carbonyl group, and the alpha-carbonyl hydrogens. Initially, we treated indolizine 5aa with NBS in acetonitrile, and we obtained a mixture from which the major isolated product was 7 (resulting from bromination of the cyclohexane moiety at the benzylic position), in 32% yield. Other attempts to brominate the indolizine core were unsuccessful. When 5aa was submitted to classical Vilsmeier–Haack formylation conditions, the stable chloro dialdehyde 830 was obtained in 70% yield after chromatographic purification. We also decided to evaluate the ability of tricyclic indolizine ketone 5aa to participate in a condensation reaction. Thus, a methanolic solution of 5aa was treated with KOH and with 2-amino-3-pyridinecarbaldehyde 9 at rt to give the new polycyclic 1,8-naphthiridine 10 in 62% yield after chromatographic purification (Scheme 7).
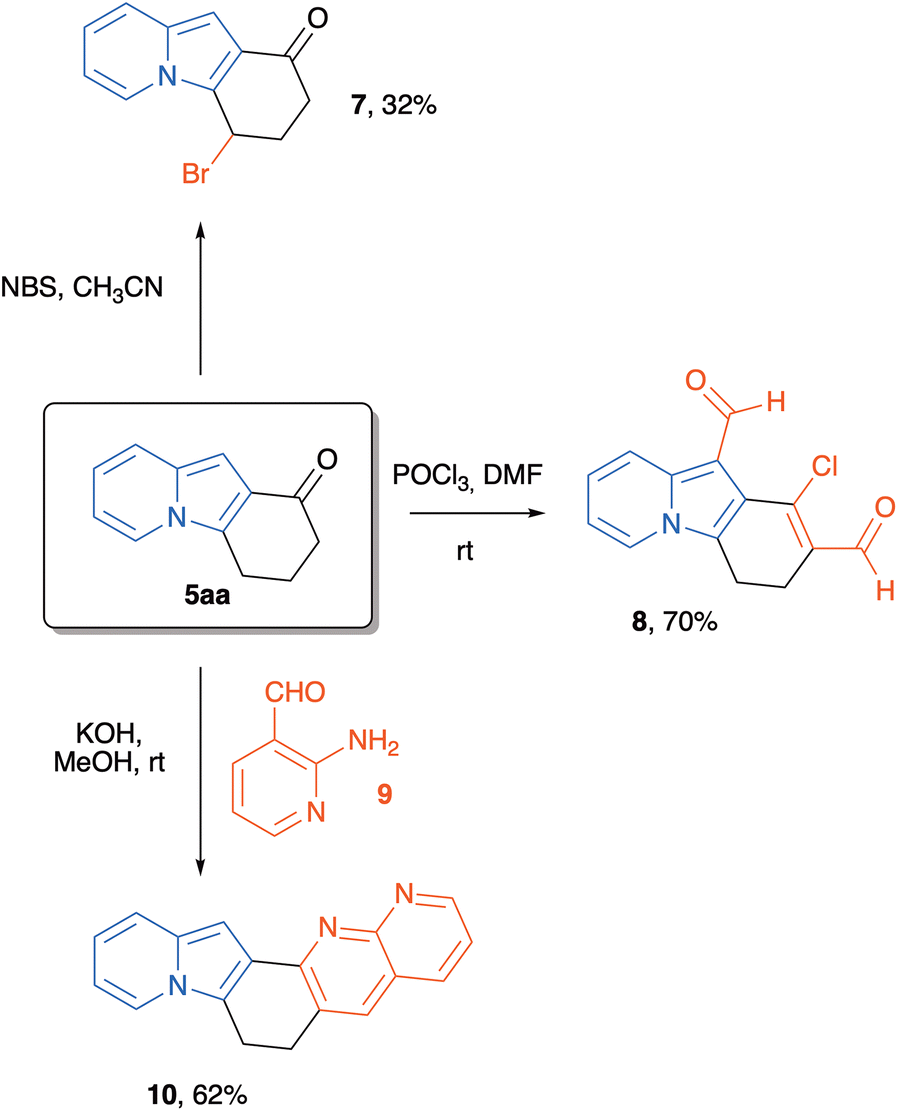 | ||
| Scheme 7 Synthetic exploration of the tricyclic indolizine 5aa. | ||
To further explore the chemical potentiality of these tricyclic indolizines, we treated 5aa with NaBH4 in methanol at rt to afford the corresponding benzylic alcohol in almost quantitative yield, after 15 min. The racemic alcohol 11 was pure enough to avoid any additional purification step. This result, associated with the structural difference between the substituents of the carbonyl group stimulated us to evaluate the behavior of this compound when treated with the Corey–Bakshi–Shibata (CBS) catalyst and borane.31 So, 5aa was reduced with the (S)-CBS reagent (at the 1 g scale) to provide the corresponding alcohol (R)-11 in 95% yield (Scheme 8). This compound was determined to have a 93% e.e., as analyzed by HPLC with a chiral stationary phase (Chiralpak® IA column).
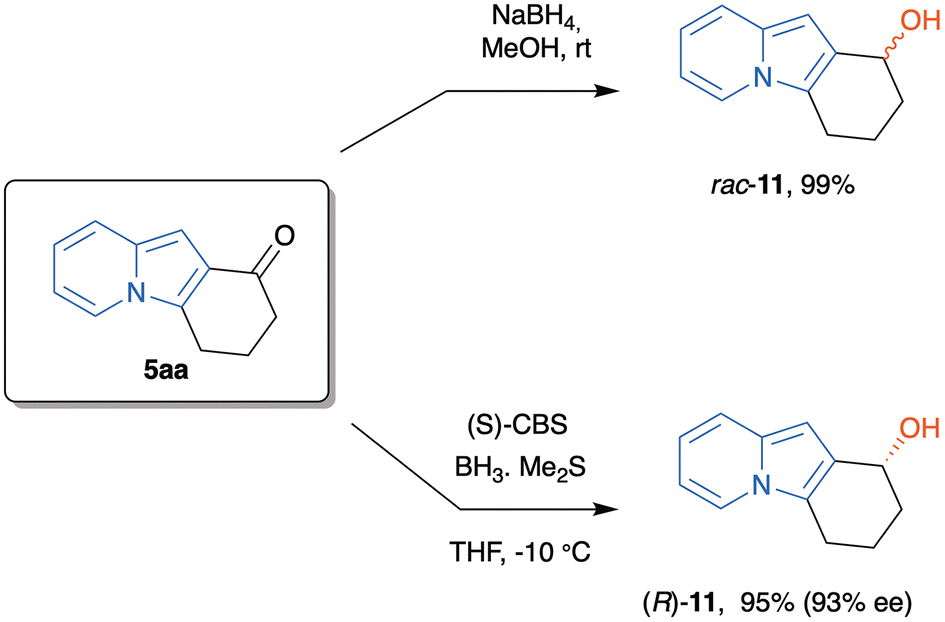 | ||
| Scheme 8 Racemic and asymmetric carbonyl reduction of the tricyclic indolizine 5aa. | ||
This set of simple chemical transformations demonstrate unambiguously the potentiality of these tricyclic indolizines as platforms for the synthesis of compounds with new carbon arrangements.
Conclusions
In summary, we have developed a straightforward approach for the synthesis of tricyclic indolizines, based on aqueous Morita–Baylis–Hillman (MBH) reactions between 2-pyridinecarbaldehydes and cyclic enones, organocatalyzed by a readily available, bi-functional Lewis base (BIA). This is the first systematic study of an organocatalyzed method for the synthesis of this class of heterocycles, starting from simple building blocks. In a single step, it was possible to prepare a set of tricyclic indolizines in isolated yields ranging from 16% to up 70%, in aqueous solution at 60 °C. The catalytic role of the bicyclic imidazolyl alcohol is restricted to the initial formation of the MBH adducts, that when generated in water can cyclize to give a zwitterionic intermediate that upon dehydration gives rise to the indolizine compounds. The facility of the cyclization strongly depends on the size of the cycloalkenone ring: while adducts derived from six-, seven- or even eight-membered cycloenones are readily transformed into the corresponding indolizines, cyclopentenone-derived MBH adducts do not cyclize. Strongly electron-withdrawing substituents, particularly when located in position 5 of the 2-pyridincarboxaldehyde, also difficult the aza-Michael cyclization step, probably due to the diminished nucleophilicity of the pyridine nitrogen. Some chemical transformations were performed on these novel heterocycles to demonstrate their synthetic usefulness.Theoretical calculations were performed to rationalize the reason why the cyclization step fails when cyclopentenones were employed. The calculations demonstrated that the transition state energies for the intramolecular aza-Michael step, that is disfavored according to Baldwin's ring-closure rules and that leads to a zwitterionic intermediate, (a) are substantially lower in water solution than in the gas-phase, and (b) are much higher for MBH adducts derived from five-membered cycloenones than for those derived from 6- and 7-membered cycloenones.
Experimental
General methods
All chemicals were used as purchased, unless otherwise noticed. Reaction progress was monitored by thin layer chromatography on silica gel (aluminum foils) and spotted under UV light (254 nm), followed by staining with ethanolic 25% phosphomolybdic acid solution, aqueous KMnO4 or vanillin in acetic acid. Purification by column chromatography was carried out with silica gel (70–230 or 230–400 Mesh). The non-commercial enones 3d, 3e, and 3f were prepared according to Yamamoto et al.26 Enone 3g was prepared according to List et al.271H NMR spectra were acquired at 600, 500, 400 or 250 MHz; 19F NMR spectra at 471 or 235 MHz; and 13C NMR spectra at 152, 126, 100 or 62.5 MHz, in CDCl3, CD2Cl2 or C6D6, at room temperature. Chemical shifts (δ) were reported in ppm and were calibrated against tetramethylsylane or the solvent residual signal according to the literature. The coupling constants (J) are reported in Hertz (Hz) and the signal multiplicity was assigned as s (singlet), d (doublet), t (triplet), dd (double doublet), dt (double triplet), td (triple doublet), tt (triple triplet), ddd (double double doublet), dddd (double double double doublet), quartet (q), multiplet (m) and broad singlet (br).
The high-resolution mass spectrometric analyses (HRMS) were performed in a Q-Tof instrument, equipped with an ESI ionization source operating in the positive mode (ESI(+)-MS). The samples were injected by direct infusion in a 40 μL min−1 flow. The following parameters were used: 3 kV capillary voltage, 20 V cone voltage, source temperature of 120 °C and nebulization gas flow of 0.5 L h−1. Before every analysis, the instrument was calibrated with an H3PO4 solution (0.005% in H2O/CH3CN 1:1) from m/z 100 to 1000. Melting points were obtained using an Electrothermal equipment model 9100 and are corrected. Compounds were named according to IUPAC rules using the program MarvinSketch 17.8.
Computational calculations
Conformational search calculations for all species were carried out in water by applying the analytical linearized Poisson–Boltzmann (ALPB)32 solvent model to find the lowest energy conformers within calculated energies below than 10 kcal mol−1 at the GFN2-xTB33 level using CREST 2.11 software. The resulting conformers were reoptimized using Gaussian 16 Rev C.0134 at the M06-2X/aug-cc-pVTZ level and using the IEFPCM35 implicit solvent model with parameters of water. Frequency calculations at the same level of the optimizations were carried out to confirm the converged geometries as either minima (without negative frequencies) or transition states (one negative frequency). A benchmarking study was carried out by calculating single point energies using several DFT functionals and the DLPNO-CCSD(T)/def2-TZVP level as the reference. DLPNO-CCSD(T)/def2-TZVP single point calculations were run using ORCA 5.0.1 package of programs.36 The ωB97xD/aug-cc-pVTZ showed the best performance when compared to the DLPNO-CCSD(T)/def2-TZVP level, being all optimization and frequency calculations repeated at this DFT level. Intrinsic reaction coordinate (IRC) calculations37 at the ωB97xD/aug-cc-pVTZ level were carried out to assess whether the obtained transition states were connected with two minima.Synthetic procedures and characterization
General procedure for the synthesis of indolizines
To a 5 mL round-bottomed flask were added 1.0 mmol of the pyridinecarbaldehyde 2, 2.0 mmol of the α,β-unsaturated cyclic ketone 3 (2.0 equiv.), 0.65 mmol of the BIA catalyst 1 (170 mg, 0.65 equiv.), 0.1 mmol of sodium dodecylsulfate (0.1 equiv., 28.8 mg) and 0.5 mL of distilled water (2 M concentration with respect to aldehyde). The flask was then sealed with a rubber septum and was magnetically stirred at 60 °C. After 12 h, the solvent was evaporated, the remaining wet solid was extracted with dichloromethane and purified using flash column chromatography (SiO2) eluting with a gradient of Hex:AcOEt (95:05–60:40) to give the desired indolizine (5), or the corresponding MBH adduct (4), or the aldol adduct (6).
Synthesis of 4-bromo-1H,2H,3H,4H-pyrido[1,2-a]indol-1-one (7)
In a flame-dried 5 mL round-bottomed flask, a solution of indolizine 5aa (185.2 mg, 1.0 mmol, 1 equiv.) in anhydrous CH3CN (1 mL) was prepared under nitrogen atmosphere. Then, freshly recrystallized N-bromosuccinimide (178.0 mg, 1.0 mmol, 1 equiv.) was added to the flask under magnetic stirring. The reaction mixture was stirred for 2 hours until complete conversion of starting material, as assessed by TLC analysis. The solvent was removed under reduced pressure, and the residue was directly purified by flash column chromatography (SiO2) eluting with hexane:ethyl acetate (95:5) to give 7 in 32% yield (84.5 mg) as a viscous brown oil. 1H NMR (500 MHz, C6D6) δ 6.92 (s, 1H), 6.84 (d, J = 9.2 Hz, 1H), 6.65 (dd, J = 7.2, 1.0 Hz, 1H), 6.20–6.13 (m, 1H), 5.96–5.89 (m, 1H), 4.43–4.38 (m, 1H), 2.42 (ddd, J = 16.0, 9.7, 5.0 Hz, 1H), 1.90–1.84 (m, 1H), 1.79 (ddd, J = 16.0, 5.0, 3.0 Hz, 1H), 1.76–1.69 (m, 1H). 13C NMR (126 MHz, C6D6) δ 187.2, 133.5, 129.7, 122.2, 121.1, 120.8, 117.9, 112.0, 97.5, 51.0, 32.1, 18.0. HRMS (ESI): m/z calcd for C12H11BrNO [M + H]+: 264.0019, found: 264.0014.
Synthesis of 1-chloro-3,4-dihydropyrido[1,2-a]indole-2,10-dicarbaldehyde (8)
In a flame-dried 25 mL round-bottomed flask, phosphorous oxychloride (76.0 μL, 0.81 mmol) was added to anhydrous DMF (2.70 mL, 0.1 M) at 0 °C under a nitrogen atmosphere. After being stirred for 1 h indolizine 5aa (50.0 mg, 0.27 mmol, 1.0 equiv.) was added and the mixture was stirred for 15 min at 0 °C. The reaction was then heat at 60 °C for 24 h. After cooling, the mixture was quenched with saturated NaHCO3 and the product was extracted with CH2Cl2 (3 × 10 mL). The combined organic fraction were washed with water (10 mL) and brine (10 mL), dried over Na2SO4 and evaporated. The residue was purified by flash column chromatography (SiO2) eluting with hexane:ethyl acetate (60:40) to give 8 in 70% yield (181.8 mg) as a viscous yellow oil. 1H NMR (500 MHz, CDCl3) δ 10.76 (s, 1H), 10.36 (s, 1H), 8.55 (d, J = 9.0 Hz, 1H), 7.84 (d, J = 6.8 Hz, 1H), 7.23 (dd, J = 9.1, 6.6 Hz, 1H), 6.97 (t, J = 6.8 Hz, 1H), 3.15–2.83 (m, 4H). 13C NMR (126 MHz, CDCl3) δ 189.5, 185.7, 142.7, 137.1, 129.5, 127.2, 125.4, 122.6, 122.3, 121.8, 115.7, 22.3, 18.8. HRMS (ESI): m/z calcd for C14H11ClNO2 [M + H]+: 260.0473, found: 260.0469.
Synthesis of 6,7-dihydropyrido[1′,2′:1,2]indolo[4,5-b][1,8]naphthyridine (10)
In a 5 mL round-bottomed flask, 5aa (185.2 mg, 1.0 mmol, 1 equiv.), 2-amino-3-pyridinecarboxaldehyde 9 (122.1 mg, 1 mmol, 1 equiv.) and MeOH (1 ml) were added sequentially. The solution was magnetically stirred, and solid KOH (112 mg, 2.0 mmol, 2 equiv.) was added in a single portion. The mixture was then heated to 40 °C until complete consumption of the starting material by TLC. After the reaction was complete, the solvent was evaporated, and the mixture was redissolved in dichloromethane. Water was then added (50 mL), and the aqueous phase was extracted with CH2Cl2 (3 × 50 mL). The combined organic layers were dried over Na2SO4, filtered, and evaporated. The crude was then purified using flash column chromatography (SiO2) eluting with hexane:ethyl acetate (50:50) to give 10 in 62% yield (168.2 mg) as a yellow solid, mp >200 °C (decomp.). 1H NMR (500 MHz, CDCl3) δ 8.97 (dd, J = 4.2, 2.0 Hz, 1H), 8.01 (dd, J = 8.0, 1.9 Hz, 1H), 7.84 (t, J = 1.2 Hz, 1H), 7.65 (ddd, J = 7.1, 2.1, 1.0 Hz, 1H), 7.43 (dt, J = 9.1, 1.1 Hz, 1H), 7.33 (dd, J = 8.0, 4.3 Hz, 1H), 7.29 (s, 1H), 6.64 (ddd, J = 9.1, 6.4, 1.0 Hz, 1H), 6.55–6.48 (m, 1H), 3.35 (t, J = 7.1 Hz, 1H), 3.14 (t, J = 7.3 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 156.3, 156.1, 152.4, 136.1, 134.0, 134.0, 130.5, 125.6, 124.1, 121.8, 121.8, 120.9, 120.6, 117.0, 111.3, 96.0, 28.8, 19.7. HRMS (ESI): m/z calcd for C18H14N3 [M + H]+: 272.1182, found: 272.1188.
Reduction of 5aa using NaBH4 (rac-11)
A solution of indolizine 5aa (185.2 mg, 1.0 mmol, 1 mmol) in methanol (10 mL) was prepared in a 25 mL round-bottomed flask. While kept under magnetic stirring, NaBH4 (75.7 mg, 2.0 mmol, 2 equiv.) was added in a single portion. After checking the total consumption of the starting material (TLC), the solvent was removed under reduced pressure, redissolved in diethyl ether (50 mL) and extracted with aq. saturated NH4Cl (1 × 10 mL), distilled water (2 × 10 mL) and brine (1 × 10 mL). The organic layer was then dried over Na2SO4, filtered, and evaporated under reduced pressure to provide the racemic alcohol 11 as a colorless solid in 99% yield (185.4 mg), mp: >100 °C (decomp.). 1H NMR (600 MHz, CD2Cl2) δ 7.67 (ddd, J = 7.0, 2.0, 1.1 Hz, 1H), 7.37 (dt, J = 9.0, 1.1 Hz, 1H), 6.69–6.63 (m, 1H), 6.54 (ddd, J = 7.0, 6.5, 1.3 Hz, 1H), 6.45 (s, 1H), 4.98 (t, J = 5.0 Hz, 1H), 2.78 (dt, J = 15.6, 6.0 Hz, 1H), 2.73–2.65 (m, 1H), 2.22–2.12 (m, 1H), 2.11–2.03 (m, 1H), 1.99–1.93 (m, 1H), 1.87 (dddd, J = 13.1, 8.3, 5.7, 2.7 Hz, 1H). 13C NMR (151 MHz, CD2Cl2) δ 132.0, 126.6, 121.6, 120.5, 118.9, 115.8, 109.7, 95.8, 64.9, 33.2, 21.0, 19.0. HRMS (ESI): m/z calcd for C12H14NO [M + H]+: 188.1070, found: 188.1079.Synthesis of enantiomerically enriched ((R)-11)
A solution of (S)-(−)-2-methyl-CBS-oxazaborolidine (277.1 mg, 1.0 mmol, 0.2 equiv.) in anhydrous THF (20 mL) was cooled to −10 °C under N2 atmosphere. To this solution, using a syringe pump (2 mL per h flow), indolizine 5aa (926 mg, 5.0 mmol, 1 equiv.) in anhydrous THF (1 mL) was added for 30 minutes. After the addition was complete, the reaction was warmed to rt, was diluted with diethyl ether (100 mL) and was extracted with aq. saturated NH4Cl (3 × 50 mL), distilled water (2 × 50 ml) and saturated NaCl (1 × 50 mL). The organic layer was then dried over Na2SO4, filtered through a pad of silica gel using ethyl acetate and evaporated to provide enantiomerically enriched alcohol (R)-11 as a colorless solid in 90% yield (168.5 mg) and 93% ee determined by HPLC (Chiralpak® IA column, eluting with hexane:2-propanol (95:05), 25 °C, 1 mL min−1). [α]D = −17 (c = 0.8, CHCl3). Spectral data were identical to those determined for the racemic compound.
Author contributions
Conceptualization, funding acquisition, supervision: AM and FC; investigation: LAZ, LVA, MTR and HS (experimental), RAC and JCP (computational calculations); writing – original draft: all authors; writing – review and editing: AM.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the São Paulo Foundation Science (Fapesp #2018/02611-0 and #2013/07600-3, to FC) and MCIN/AEI/10.13039/501100011033 and ERDF funds (PID2020-116846GB-C21, to AM) for financial support. LAZ (#2013/24386-5 and BEPE #2014/26027-5), HS (#2015/09205-0) and RAC (#2015/00975-7) thank Fapesp for fellowships. FC is also grateful to the Brazilian National Council for Scientific and Technological (CNPq) for financial support and research fellowship (307840/2014-0 and 301330/2018-2). LVA thanks CAPES for her fellowship. JCP is grateful to MCIN/AEI and ERDF for the “María de Maeztu” grant CEX2021-001202-M.References
- (a) A. P. Taylor, R. P. Robinson, Y. M. Fabian, D. C. Blakemore, L. H. Jones and O. Fadeyl, Org. Biomol. Chem., 2016, 14, 6611–6637 RSC; (b) E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- (a) B. Szefler, P. Czelen and M. V. Diudea, Curr. Comput.-Aided Drug Des., 2017, 13, 22–29 CrossRef CAS PubMed; (b) C. Wang, H. Jia, Z. Li, H. Zhang and B. Zhao, RSC Adv., 2016, 6, 21814–21821 RSC.
- (a) J. P. Michael, Alkaloids: Chem. Biol. Perspect., 2016, 75, 1–498 CAS; (b) J. Zhang, S. L. Morris-Natschke, D. Ma, X.-F. Shang, C.-J. Yang, Y.-Q. Liu and K. H. Lee, Med. Res. Rev., 2021, 41, 928–960 CrossRef CAS PubMed.
- (a) S.-H. Moon, Y. Jung, S. H. Kim and Y. Kim, Bioorg. Med. Chem. Lett., 2016, 26, 110–113 CrossRef CAS PubMed; (b) A. Ghinet, C.-M. Abuhaie, P. Gautret, B. Rigo, J. Dubois, A. Farce, D. Belei and E. Bîcu, Eur. J. Med. Chem., 2015, 89, 115–127 CrossRef CAS PubMed; (c) A. Boot, A. Brito, T. van Wezel, H. Morreau, M. Costa and F. Proenca, Anti-Cancer Res., 2014, 34, 1673–1677 CAS.
- (a) E. Faghih-Mirzaei, M. Seifi, M. Abaszadeh, K. Zomorodian and H. Helali, Iran. J. Pharm. Res., 2018, 17, 883–895 CAS; (b) A.-M. Olaru, V. Vasilachu, R. Danae and I. I. Mangalagiu, J. Enzyme Inhib. Med. Chem., 2017, 32, 1291–1298 CrossRef CAS PubMed; (c) R. Danac, C. M. Al Matarneh, S. Shova, T. Daniloaia, M. Balan and I. I. Mangalagiu, Bioorg. Med. Chem., 2015, 23, 2318–2327 CrossRef CAS PubMed; (d) Y.-M. Shen, P.-C. Lv, W. Chen, P.-G. Liu, M.-Z. Zhang and H.-L. Zhu, Eur. J. Med. Chem., 2010, 45, 3184–3190 CrossRef CAS PubMed; (e) L.-L. Gundersen, A. H. Negussie and F. Rise, Eur. J. Pharm. Sci., 2007, 30, 26–35 CrossRef CAS PubMed; (f) L.-L. Gundersen, K. E. Malterud and A. H. Negussie, Bioorg. Med. Chem., 2003, 11, 5409–5419 CrossRef CAS PubMed.
- S. Shrivastava, P. Shrivastava, R. Bandesh, P. N. Tripathi and A. Tripathi, Bioorg. Med. Chem., 2017, 25, 4424–4432 CrossRef CAS PubMed.
- S. Chen, Z. Xia, M. Nagai, R. Lu, E. Kostik, T. Przewloka, M. Song, D. Chimmanamada, D. James, S. Zhang, J. Jiang, M. Ono, K. Koya and L. Sun, MedChemComm, 2011, 2, 176–180 RSC.
- Y. Xue, J. Tang, X. Ma, Q. Li, B. Xie, Y. Hao, H. Jin, K. Wang, G. Zhang, L. Zhang and L. Zhang, Eur. J. Med. Chem., 2016, 115, 94–108 CrossRef CAS PubMed.
- (a) V. Sharma and V. Kumar, Med. Chem. Res., 2014, 23, 3593–3606 CrossRef CAS; (b) G. M. Cingolani, F. Claudi and F. Venturi, Eur. J. Med. Chem., 1988, 23, 291–294 CrossRef CAS; (c) I. Antonini, F. Claudi, U. Gulini, L. Micossi and F. Venturi, J. Pharm. Sci., 1979, 68, 321–324 CrossRef CAS PubMed; (d) M. Cardellini, F. Claudi, M. Grifantini, U. Gulini and S. Martelli, J. Pharm. Sci., 1977, 66, 259–262 CrossRef CAS PubMed; (e) F. Claudi, M. Grifantini, U. Gulini, S. Martelli and P. Natalini, J. Pharm. Sci., 1977, 66, 1355–1357 CrossRef CAS PubMed; (f) I. Antonini, M. Cardellini, F. Claudi, P. Franchetti, U. Gulini, G. De Caro and F. Venturi, J. Pharm. Sci., 1977, 66, 1692–1696 CrossRef CAS PubMed.
- (a) R. Sarkar, T. Chaudhuri, A. Karmakar and C. Mukhopadhyay, Org. Biomol. Chem., 2015, 13, 11674–11686 RSC; (b) E. Kim, Y. Lee and S. B. Park, Acc. Chem. Res., 2015, 48, 538–547 CrossRef CAS PubMed; (c) E. J. Choi, E. Kim, Y. Lee, A. Jo and S. B. Park, Angew. Chem., Int. Ed., 2014, 53, 1346–1350 CrossRef CAS PubMed; (d) M. F. Z. J. Amaral, L. A. Deliberto, C. R. de Souza, R. M. Z. G. Naal, Z. Naal and G. C. Clososki, Tetrahedron, 2014, 70, 3249–3258 CrossRef CAS; (e) B. Lui, Z. Wang, N. Wu, M. Li, J. You and J. Lan, Chem. – Eur. J., 2012, 18, 1599–1603 CrossRef PubMed; (f) E. Kim, M. Koh, B. J. Lim and S. P. Park, J. Am. Chem. Soc., 2011, 133, 6642–6649 CrossRef CAS PubMed; (g) E. Kim, S. Lee and S. B. Park, Chem. Commun., 2011, 47, 7734–7736 RSC.
- J. H. Delcamp, A. Yella, T. W. Holcombe, M. K. Nazeeruddin and M. Grätzel, Angew. Chem., Int. Ed., 2013, 52, 376–380 CrossRef CAS PubMed.
- (a) M. Scholtz, Ber. Dtsch. Chem. Ges., 1912, 45, 734–746 CrossRef CAS; (b) V. Boelkelheide and R. J. Windgassen, J. Am. Chem. Soc., 1959, 81, 1456–1459 CrossRef. For a review, see: (c) T. Uchide and K. Matsumoto, Synthesis, 1976, 209–236 CrossRef.
- A. E. Tschitschibabin, Ber. Dtsch. Chem. Ges., 1912, 60, 1607–1617 CrossRef.
- Selected reviews: (a) S. C. K. N. Venugopala, M. A. Khedr, M. Attimarad, B. Padmashali, R. S. Kulkarni, R. Venugopala and B. Odhav, J. Basic Clin. Pharm., 2017, 8, 49–60 Search PubMed; (b) C. R. de Souza, A. C. Gonçalves, M. F. Z. J. Amaral, A. A. dos Santos and G. C. Clososki, Targets Heterocycl. Chem., 2017, 20, 365–392 Search PubMed; (c) K. M. Elattar, I. Youssef and A. A. Fadda, Synth. Commun., 2016, 46, 719–744 CrossRef CAS; (d) G. S. Singh and E. E. Mmatli, Eur. J. Med. Chem., 2011, 46, 5237–5257 CrossRef CAS PubMed. For some alternative methods for the synthesis of indolizines, see: (e) A. R. Hardin and R. Sarpong, Org. Lett., 2007, 9, 4547–4550 CrossRef CAS PubMed; (f) X.-C. Tan, Y. Laing, F.-P. Bao, H.-S. Wang and Y.-M. Pan, Tetrahedron, 2014, 70, 6717–6722 CrossRef CAS; (g) S. Park and I. Kim, Tetrahedron, 2015, 71, 1982–1991 CrossRef CAS; (h) B. Sahoo, M. N. Hopkinson and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 15545–15549 CrossRef CAS PubMed; (i) X. Li, J. Zhao, X. Xie and Y. Liu, Org. Biomol. Chem., 2017, 15, 8119–8133 RSC.
- For reviews, see: (a) B. Sadowski, J. Klajn and D. T. Gryko, Org. Biomol. Chem., 2016, 14, 7804–7828 RSC; (b) J. Hui, Y. Ma, J. Zhao and H. Cao, Org. Biomol. Chem., 2021, 19, 10245–10258 RSC.
- (a) Y. Liu, H.-Y. Hu, Q.-J. Liu, H.-W. Hu and J.-H. Xu, Tetrahedron, 2007, 63, 2024–2033 CrossRef CAS; (b) W. M. Bloch, S. M. Derwent-Smith, F. Issa, J. C. Morris, L. M. Redina and C. J. Sumby, Tetrahedron, 2011, 67, 9368–9375 CrossRef CAS; (c) N. Chernyak, D. Tilly, Z. Li and V. Gevorgyan, ARKIVOC, 2011, v, 76–91 Search PubMed; (d) M. Kim, Y. Jung and I. Kim, J. Org. Chem., 2013, 78, 10395–10404 CrossRef CAS PubMed; (e) Y. Shi and V. Gevorgyan, Chem. Commun., 2015, 51, 17166–17169 RSC; (f) P. N. Bagle, M. V. Mane, K. Vanka, D. R. Shinde, S. R. Shaikh, R. Gonnade and N. T. Patil, Chem. Commun., 2016, 52, 14462–14465 RSC; (g) T. Lepitre, R. Le Biannic, M. Othman, A. M. Lawson and A. Daïch, Org. Lett., 2017, 19, 1978–1981 CrossRef CAS PubMed; (h) S. R. Pathipati, A. van der Werf and N. Selander, Org. Lett., 2018, 20, 3691–3694 CrossRef CAS PubMed.
- (a) K. Morita, Z. Suzuki and H. Hirose, Bull. Chem. Soc. Jpn., 1968, 41, 2815 CrossRef CAS; (b) A. B. Baylis and M. E. D. Hillman, German Patent, 2155113, 1972 Search PubMed. For reviews on the Morita-Baylis-Hillman reaction, see: (c) M. Shi, F.-J. Wang, M.-X. Zhao and Y. Wei, The Chemistry of the Morita-Baylis-Hillman Reaction, RSC Publishing, Cambridge, UK, 2011 RSC; (d) T. N. Reddy and V. J. Rao, Tetrahedron Lett., 2018, 59, 2859–2875 CrossRef; (e) M. S. Santos, F. Coelho, C. G. Lima-Junior and M. L. A. A. Vasconcellos, Curr. Org. Synth., 2015, 12, 830–852 CrossRef CAS; (f) S. Bhowmik and S. Batra, Curr. Org. Chem., 2014, 18, 3078–3119 CrossRef CAS; (g) Y. Wei and M. Shi, Chem. Rev., 2013, 113, 6659–6690 CrossRef CAS PubMed; (h) D. Basavaiah and D. G. Veeraraghavaiah, Chem. Soc. Rev., 2012, 41, 68–78 RSC; (i) T.-Y. Lui, M. Xie and Y.-C. Chen, Chem. Soc. Rev., 2012, 41, 4101–4112 RSC; (j) D. Basavaiah, S. B. Reddy and S. S. Badsara, Chem. Rev., 2010, 110, 5447–5674 CrossRef CAS PubMed; (k) R. O. M. A. Souza and L. S. M. Miranda, Mini-Rev. Org. Chem., 2010, 7, 212–220 CrossRef; (l) G.-N. Ma, J.-J. Jiang, M. Shi and Y. Wei, Chem. Commun., 2009, 5496–5514 RSC; (m) V. Singh and S. Batra, Tetrahedron, 2008, 64, 4511–4574 CrossRef CAS; (n) Y.-L. Shi and M. Shi, Eur. J. Org. Chem., 2007, 2905–2916 CrossRef CAS; (o) D. Basavaiah, K. V. Rao and R. J. Reddy, Chem. Soc. Rev., 2007, 36, 1581–1588 RSC; (p) W. P. Almeida and F. Coelho, Quim. Nova, 2000, 23, 98–101 CrossRef; (q) E. Ciganek, Org. React., 1997, 51, 201–350 CAS.
- (a) D. Basavaiah, B. Lingaiah, G. C. Reddy and B. C. Sahu, Eur. J. Org. Chem., 2016, 2398–2403 CrossRef CAS; (b) B. V. M. Teodoro, J. T. M. Correia and F. Coelho, J. Org. Chem., 2015, 80, 2529–2538 CrossRef CAS PubMed; (c) D. Basavaiah, G. Veeraraghavaiah and S. S. Badsara, Org. Biomol. Chem., 2014, 12, 1551–1555 RSC; (d) S. M. D. Cunha, R. G. de Oliveira and M. L. A. A. Vasconcellos, J. Braz. Chem. Soc., 2013, 24, 432–438 CrossRef CAS; (e) H. Zhu, N. Shao, T. Chen and H. Zou, Chem. Commun., 2013, 49, 7738–7740 RSC; (f) V. Singh, S. Hutait and S. Batra, Eur. J. Org. Chem., 2010, 3684–3691 CrossRef CAS; (g) D. Basavaiah, B. Devendar, D. V. Lenin and T. Satyanarayaana, Synlett, 2009, 411–416 CrossRef CAS; (h) M. Jiang, Q. He, C. Yang and Y. Xie, Heterocycles, 2008, 75, 2659–2666 CrossRef CAS; (i) S. Sun, M. Wang, H. Deng and Q. A. Liu, Synthesis, 2008, 573–583 CAS; (j) D. Basavaiah and A. J. Rao, Tetrahedron Lett., 2003, 44, 4365–4368 CrossRef CAS; (k) D. Basavaiah and A. J. Rao, Chem. Commun., 2003, 604–605 RSC.
- For reviews on the organocatalyzed synthesis of polycyclic systems, see: (a) A. Moyano and R. Rios, Chem. Rev., 2011, 111, 4703–4832 CrossRef CAS PubMed; (b) Y. Wang and D.-M. Du, Org. Chem. Front., 2020, 7, 3266–3283 RSC; (c) M.-S. Tu, K.-W. Chen, P. Wu, Y.-C. Zhang, X.-Q. Liu and F. Shi, Org. Chem. Front., 2021, 8, 2643–2672 RSC; (d) H.-H. Zhang and F. Shi, Acc. Chem. Res., 2022, 55, 2562–2580 CrossRef CAS PubMed; (e) W. Tan, J.-Y. Zhang, C.-H. Gao and F. Shi, Sci. China: Chem., 2023, 66, 966–992 CrossRef CAS.
- J. C. Gomes, M. T. Rodrigues Jr., A. Moyano and F. Coelho, Eur. J. Org. Chem., 2012, 6861–6866 CrossRef CAS.
- J. C. Gomes, J. Sirvent, M. T. Rodrigues Jr., A. Moyano and F. Coelho, Org. Lett., 2013, 15, 5838–5841 CrossRef CAS PubMed.
- P. N. Joshi, L. Purushottam, N. K. Das, S. Mukherjee and V. Rai, RSC Adv., 2016, 6, 208–211 RSC.
- L. Raich, H. Santos, J. C. Gomes, M. T. Rodrigues Jr., R. Galaverna, M. N. Eberlin, F. Coelho, C. Rovira and A. Moyano, ACS Catal., 2018, 8, 1703–1714 CrossRef CAS.
- K.-S. Park, J. Kim, H. Choo and Y. Chong, Synlett, 2007, 395–398 CAS.
- (a) P. M. Weintraub, P. L. Tiernan and J. C. Huffman, J. Heterocycl. Chem., 1987, 24, 561–563 CrossRef CAS; (b) Z. Zhang, F. Xie, J. Jia and W. Zhang, J. Am. Chem. Soc., 2010, 132, 15939–15941 CrossRef CAS PubMed.
- B. Maji and H. Yamamoto, J. Am. Chem. Soc., 2015, 137, 15957–15963 CrossRef CAS PubMed.
- O. Lifchits, M. Mahlau, C. M. Reisinger, A. Lee, C. Farès, I. Polyak, G. Gopakumar, W. Thiel and B. List, J. Am. Chem. Soc., 2013, 135, 6677–6693 CrossRef CAS PubMed.
- See ref. 23 for a detailed experimental and computational study of the mechanism of the catalysis of the MBH reaction by the bicyclic imidazolyl alcohol 1.
- J. E. Baldwin, J. Chem. Soc., Chem. Commun., 1976, 734–736 RSC.
- For a similar transformation, see: M. Layek, M. A. Reddy, A. V. D. Rao, M. Alvala, M. K. Arunasree, A. Islam, K. Mukkanti, J. Iqbal and M. Pal, Org. Biomol. Chem., 2011, 9, 1004–1007 RSC.
- (a) E. J. Corey, R. K. Bahski and S. Shibata, J. Am. Chem. Soc., 1987, 109, 5551–5553 CrossRef CAS; (b) E. J. Corey, S. Shibata and R. K. Bakshi, J. Org. Chem., 1988, 53, 2861–2863 CrossRef CAS.
- S. Ehlert, M. Stahn, S. Spicher and S. Grimme, J. Chem. Theory Comput., 2021, 17, 4250–4261 CrossRef CAS PubMed.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- B. Mennucci, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 386–404 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- K. Fukui, J. Phys. Chem., 1970, 74, 4161–4163 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR (1H, 13C, 19F) spectra for compounds 4ab, 4eb, 4gb, 4ja, 5aa–5ic, 6ag, 6eb, 7, 8, 10 and 11; HPLC chromatograms of compounds rac-11 and (R)-11; cartesian coordinates, Gibbs free energies and lowest harmonic vibrational frequencies (LHVFs) computed for each species at the ωB97xD/aug-cc-pVTZ level in water using the IEFPCM implicit solvent model; relative energies of two transition states calculated for different DFT functionals. See DOI: https://doi.org/10.1039/d3ob00346a |
| This journal is © The Royal Society of Chemistry 2023 |