Synthesis of heterocycles by radical cyclisation
W. Russell Bowman, Colin F. Bridge and Philip Brookes
Department of Chemistry, Loughborough University, Loughborough, Leicestershire, UK LE11 3TU
First published on UnassignedUnassigned24th December 1999
Abstract
Covering: July 1996 to June 1998.
Previous review: Contemp. Org. Synth., 1997, 4, 261.
1 Introduction
The use of radical cyclisation for the synthesis of heterocycles continues to grow and many new methodologies have been published in addition to the use of tributyltin hydride (Bu3SnH) or related triorganostannanes. The majority of radical cyclisations in heterocyclic chemistry are still carried out using Bu3SnH. The reaction conditions are generally to use an excess of Bu3SnH with a smaller equivalent (10–25 mol%) of a radical initiator, most commonly azobisisobutyronitrile (AIBN). The reactions are generally refluxed in benzene or toluene for 1–10 hours. Bu3SnH mediated reactions are well known and therefore the mechanisms will not be discussed in detail. In Section 12 at the end of the review the different reagents and methods for generating the radicals are discussed. Photochemical reactions not proceeding by chain reactions, e.g. by photochemical generation of biradicals have been excluded.Most radical cyclisations used for the syntheses of heterocycles proceed by 5-exo-trig regioselectivity. Therefore, the review has not been divided on the basis of ring size. The review has largely excluded heterocyclic syntheses in which the heterocyclic ring(s) are not part of the radical cyclisation. Therefore, carbocyclic cyclisations in molecules which contain a heterocycle are not included. A new section on macrocyclisation has been included (Section 11). Stereoselective synthesis of heterocycles has become increasingly important and a number of examples are shown.
A number of reviews which include the synthesis of heterocycles via radical cyclisation have been published. Reviews on radical chemistry which contain significant sections on heterocyclic synthesis include the use of xanthates in free radical reactions,1 the formation of C–C bonds mediated by cerium reagents, mainly cerium(IV) ammonium nitrate (CAN)![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 2 and the use of N-aziridinyl imines and azides for the generation of C- and N-centred radicals which can undergo cyclisation.3 The use of temporarily silicon-tethered molecules in synthesis,4 in particular the use of (bromomethyl)dimethylsilyl ethers, has been reviewed. Free radical mediated macrocylisations and transannular cyclisations5 and the synthesis of tetrahydro-furanyl and -pyranyl derivatives by radical cyclisation onto the β-position of β-alkoxyacrylates have also been reviewed.6
2 and the use of N-aziridinyl imines and azides for the generation of C- and N-centred radicals which can undergo cyclisation.3 The use of temporarily silicon-tethered molecules in synthesis,4 in particular the use of (bromomethyl)dimethylsilyl ethers, has been reviewed. Free radical mediated macrocylisations and transannular cyclisations5 and the synthesis of tetrahydro-furanyl and -pyranyl derivatives by radical cyclisation onto the β-position of β-alkoxyacrylates have also been reviewed.6
2 Natural product synthesis
Radical cyclisation continues to form one of the central methodologies for the synthesis of natural products containing heterocyclic rings. Complex heterocyclic systems have been constructed with increasing ingenuity in radical cyclisations. These radical cyclisation protocols commonly have several advantages over non-radical methods which require laborious multi-step alternative syntheses. The radical cyclisations which do not generally suffer from steric hindrance or racemisation problems, can be carried out in neutral organic solutions and radical cascade reactions allow the construction of two or more rings in one-pot reactions. In this section a number of illustrative syntheses are discussed while many others are detailed in respective sections defined by the nature of the heterocyclic ring(s).One of the most novel radical methodologies continues to be developed by Curran and co-workers for the synthesis of the important anticancer alkaloid camptothecin 17,8 and analogues such as mappicine.9 This cascade methodology is unusual in that a complex heteroarene system is constructed as opposed to alicyclic heterocycles. The development of this protocol indicates the potential for the use of cascade radical cyclisations for the syntheses of complex multi-ring heterocycles, a direction for synthesis which has barely been exploited at this time.
In this protocol, rings B and C are put together in a one-pot radical cascade reaction. The Curran group has now reported the syntheses of most of the camptothecin group of anticancer agents using this protocol as well as new analogues which show promising improved biological activity. The synthesis of camptothecin 1, the main alkaloid in the group, is shown in Scheme 1 as an example of this protocol. Photolysis of the N-propargyl-6-iodo-2-pyridone 2 in the presence of phenyl isocyanide and hexamethylditin generates the pyridone radical 3 which undergoes bimolecular addition to the reactive isonitrile. The new radical 4 thus generated undergoes 5-exo-dig cyclisation onto the pendant alkyne and the intermediate vinyl radical 5 cyclises onto the benzene ring. The intermediate π-radical 6 undergoes an oxidation by a mechanism which is as yet unclear to yield the pentacyclic camptothecin in good yield. The methodology has been used for a wide range of analogues with substituents on the alkyne, benzene ring A and the pyridone ring.
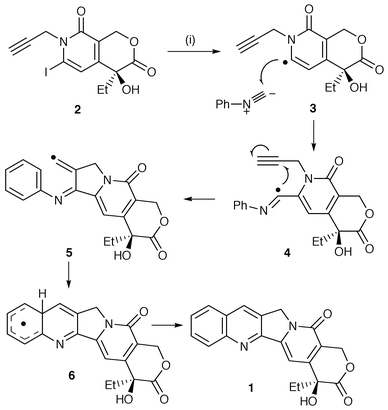 | ||
| Scheme 1 Reagents and conditions: i, PhNC, (Me3Sn)2, PhH, sun lamp, 70 °C, 8 h, 63%. | ||
An area of continuing and important research is to develop new methods of radical generation which avoid the use of the toxic triorganotin hydrides which are also troublesome to separate from reaction products. Zard and co-workers have elaborated a methodology using xanthates as radical precursors.10,11 Thermal dissociation or treatment with lauroyl peroxide as an initiator generates radicals which are able to undergo cyclisations to yield cyclised radicals which abstract the xanthate groups from the radical precursors to maintain the cycle of radical chain reactions. This protocol is illustrated in Scheme 2 for the synthesis of lactones by 5-exo cyclisation.10 An application of the protocol is also shown in Scheme 2 for the synthesis of the butenolide (±)-cinnamolide 7.10 Initial homolysis of the xanthate 8 yields acyloxy radical intermediate 9 which undergoes 5-exo cyclisation onto a suitably placed alkene. The cyclised lactone radical 10 completes the chain reaction by the abstraction of the xanthate group to generate the starting radical 9 and the xanthate product 11. Application of the procedure to starting xanthate 12 yields the lactone 13 in a stereoselective cis 5-exo cyclisation. Lactone 13 was readily transformed to the natural product 7 to complete the total synthesis. A short synthesis of tetracyclic alkaloid (±)-matrine by a radical cascade from a xanthate precursor as the key step has also been reported by Zard and co-workers.11
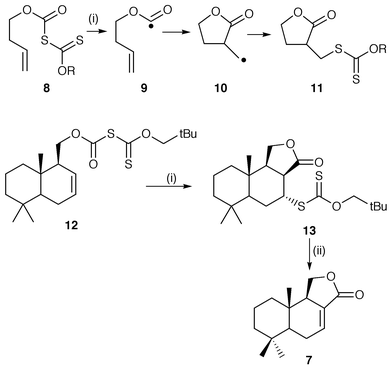 | ||
| Scheme 2 Reagents and conditions: i, hν (500 W), PhMe, 111 °C, 9.5 h, 51% from alcohol; ii, DBU, CHCl3, rt, 15 min, 80%. | ||
The use of aryl radical cyclisations to form benzoheterocycles is now a common radical procedure and is illustrated with the synthesis of the Amaryllidaceae alkaloid α-lycorane 14 (Scheme 3).12 The key step involves a 6-endo cyclisation of the intermediate aryl radical, generated from starting material 15, onto the β-position of an enamide using the standard Bu3SnH procedure. The cyclised galanthan product 16 was obtained in 79% yield as a single diastereomer. The radical synthesis of benzoheterocycles is more fully discussed in section 9.
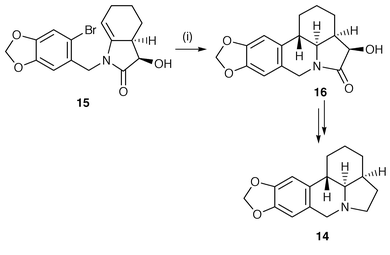 | ||
| Scheme 3 Reagents and conditions: i, Bu3SnH, AIBN, PhH, reflux, 79%. | ||
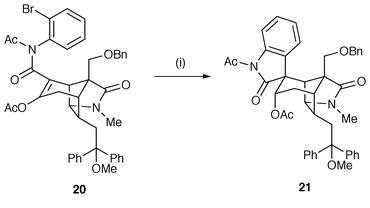
A number of syntheses of indole alkaloids have been reported using radical cyclisation. A short synthetic route to (±)-geissoschizine 17 was developed which features the construction of a corynanthe-skeleton 19via a 6-exo radical cyclisation of a vinyl radical generated from the starting indole 18 (Scheme 4).13 The 6-exo cyclisation is facilitated by use of an α,β-unsaturated ester to speed up the rate. The synthesis is also notable in the use of triethylborane (Et3B) with catalytic oxygen as the initiator which allows the reaction to be carried out at room temperature. In studies of the synthesis of the complex indole alkaloid gelsimine, radical cyclisation of the intermediate aryl radical, generated from precursor 20, yields the oxindole moiety on the gelsimine skeleton 21 in high yield (Scheme 5).14 The procedure allows the oxindole to be put together at a later stage in the synthesis by a facile route. The oxindole procedure is now well used and normally is selective for 5-exo cyclisation of the aryl radical onto the α-position of the α,β-unsaturated amide.
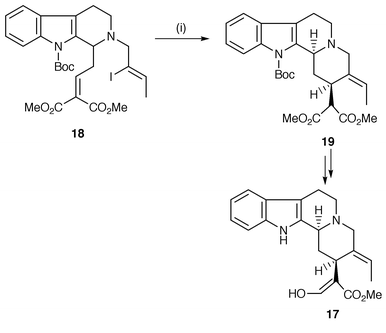 | ||
| Scheme 4 Reagents and conditions: i, Bu3SnH, Et3B, PhMe, rt, 19 (E) 33%, 19 (Z) 17%. | ||
 | ||
| Scheme 5 Reagents and conditions: i, Bu3SnH, AIBN, PhH, hν. | ||
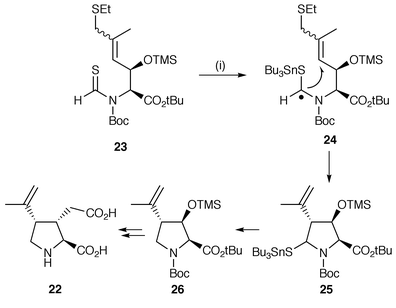
The synthesis of (−)-α-kainic acid 22 has been achieved using a novel radical protocol which involves addition of tributyltin radicals to the sulfur atom of a thioaldehyde in precursor 23 (Scheme 6).15 The resulting radicals 24 undergo stereoselective 5-exo cyclisation to yield 25. The Bu3SnS-group is reduced off during the radical reaction by further Bu3SnH to yield the trisubstituted pyrrolidine 26, which was converted to (−)-α-kainic acid. An equivalent radical cyclisation using thiols in place of Bu3SnH is reported in the same study.
 | ||
| Scheme 6 Reagents and conditions: i, Bu3SnH, AIBN, 73%. | ||
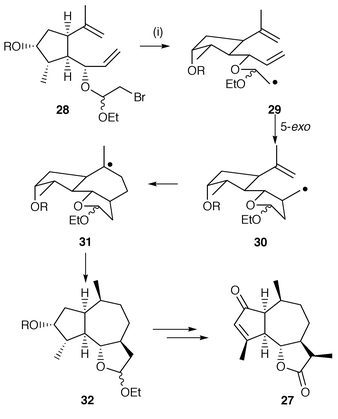
The synthesis of the sesquiterpenes, (+)-cladantholide 27 and (−)-estafiatin, which are representative of guaidanolide lactones have been synthesised using an unusual 5-exo, 7-endo cyclisation (Scheme 7).16 The cascade precursor 28 is prepared by the standard method of adding 1,2-dibromo-1-ethoxyethane to a suitable alcohol. The radical 29 generated from the β-bromoacetal undergoes normal 5-exo cyclisation using Bu3SnH. The novel reaction is the second radical cyclisation in the cascade in which the intermediate radical 30 selectively undergoes 7-endo cyclisation to yield 32via intermediate 31 to complete the 5,7-membered ring structure which is common in sesquiterpenes. In the second cyclisation, 6-exo regioselectivity is disfavoured because of the methyl substituent which creates steric hindrance. However, the selective 7-endo cyclisation is nevertheless surprising.
 | ||
| Scheme 7 Reagents and conditions: i, syringe pump addition, Bu3SnH, AIBN, PhH, reflux. | ||
3 Nitrogen heterocycles
The synthesis of nitrogen heterocycles using radical methods continues to be of increasing interest. In particular, the synthesis of pyrrolidines by 5-exo cyclisation is well suited to the use of radical cyclisation. The radical can be generated in a number of positions relative to the N-heteroatom. The first of these is the use of aminyl radicals and a number of applications have been reported. α-Amino acid aminyl radicals derived from sulfenamide precursors undergo 5-exo-trig cyclisations onto suitably placed N-alkenyl or α-alkenyl chains on the amino acids with reasonable diastereoselectivity (Scheme 8).17 The α-ester of the amino acid imparts electrophilic behaviour to the aminyl radicals and facilitates cyclisation onto alkenes. The aminyl radical precursors are prepared from the α-amino esters using benzenesulfenyl chloride in high yield. Cyclisation of aminyl radicals 34, generated from α-amino esters with N-alkenyl groups 33, i.e. with the α-ester exo to the ring, yield pyrrolidines 35 in good yields with reasonable diastereoselectivity but also yield some reduced uncyclised material (Scheme 8). When the aminyl radicals 37 are generated from α-amino esters with side chain alkenyl groups 36, proline analogues are synthesised (Scheme 8). When the primary sulfenamide 36a was used a very high yield of the respective proline 38a was obtained. However, when the N-benzyl derivative 36b was used the yield of 38b was only 44% with uncyclised reduced products. The effect of the electron donating benzyl group is sufficient to lower the electrophilicity of the aminyl radical centre and make cyclisation less favourable.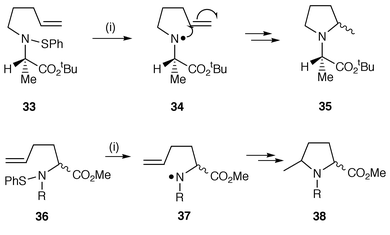 | ||
| Scheme 8 Reagents and conditions: i, Bu3SnH, AMBN, PhMe, reflux, 6 h, 81%; a. R = H 92%, de = 57%; b. R = Bn 44%. | ||
Amidyl radicals which are more electrophilic than aminyl radicals undergo cyclisation more easily. Amidyl radicals 40 generated from Bu3SnH mediated homolysis of O-benzoyl hydroxamic acid derivatives 39 undergo 4-exo-trig cyclisation to furnish β-lactam derivatives 42 together with reduced and rearrangement products (Scheme 9).18 The reaction is biased to encourage the unfavourable 4-exo cyclisation by use of a styryl side chain which yields a stable intermediate benzylic radical 41.
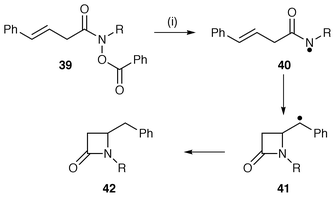 | ||
| Scheme 9 Reagents and conditions: i, Bu3SnH, AIBN, syringe pump 8 h, 1∶1 cyclohexane–toluene. | ||
Further studies of the cyclisation of aminyl radicals provide more examples of the potential for synthesis but also useful mechanistic data which will help plan syntheses.19–21 Different data indicate reversibility19,20 and irreversibility21 of 5-exo aminyl radical cyclisation. The latest study suggests that the cyclisation is reversible.20 The problems of reversibility can be overcome by use of Lewis acids to enhance the electrophilic behaviour of aminyl radicals and facilitate ready synthesis of pyrrolidines.22 The use of amidyl radicals, which are more electrophilic than aminyl radicals, for the synthesis of 5- and 6-membered ring lactams has been further developed with extensive kinetic measurements.23 Cyclisation of iminyl radicals to yield 5- and 6-membered ring nitrogen heterocycles has also been studied with measurement of the rates of cyclisation.24
A large number of radical reactions using cyclisation of radicals, β to the nitrogen atom, onto β-alkenes have been used for the synthesis of a wide range of pyrrolidines. For instance, γ-lactams have been prepared by several methods.25,26 2-Iodo-N-(prop-2-enyl)acetamides upon treatment with Et3B in boiling benzene undergo iodine atom transfer cyclisation to afford the 4-(iodomethyl)pyrrolidin-2-ones in high yields (Scheme 10).25 Triethylborane (Et3B) and oxygen were used to initiate the reaction thus providing a synthetic route which does not require Bu3SnH. In the iodine abstraction mechanism, the cyclised radical 45 abstracts iodine from the starting material 43 to yield the product iodide 46 and intermediate radical 44. The methodology was also extended to the synthesis of γ-lactones. γ-Lactams (pyroglutamates) have been synthesised from dehydroalanines which contain chiral ester auxiliaries via 5-endo radical cyclisations using Bu3SnH.26 No improvement on the cyclisation reaction was found when using triethylborane as the initiator.
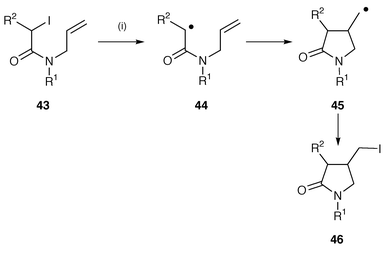 | ||
| Scheme 10 Reagents and conditions: i, Et3B, PhH, reflux; R1 = Me, Bn, Ts; R2 = H, Me. | ||
β-Lactams have been synthesised by several methods using radical cyclisation. All the methods have some factor in the mechanism to favour 4-membered ring cyclisation which is otherwise unfavourable. Zard and co-workers27 have applied their xanthate methodology to β-lactams by cyclisation of N-alkenylacetamides. An example is shown in Scheme 11. Lauroyl peroxide is used to initiate the reaction and abstract the xanthate group from starting acetamide 47. The reversible and unfavourable 4-exo cyclisation is pushed to completion by a rapid β-elimination of phenylthiyl radicals from intermediate 48 to yield the β-lactam 49. A number of similar protocols with xanthates rely on subsequent reactions in the cascade which remove the cyclised radical from the equilibrium. The protocol using xanthates is attractive because it avoids the use of the toxic and troublesome triorganotin hydrides. Zard and co-workers28 have also synthesised β-lactams from N-alkenyl-trichloroacetamides using nickel powder for generating the intermediate radicals. β-Lactams can also be synthesised using Bu3SnH from N-alkenyl-α-bromoacetamides which contain a radical stabilising aryl substituent on the β-position of the alkene.29 The stabilising substituent on the intermediate benzylic cyclised radical pushes the unfavourable equilibrium towards 4-exo cyclisation. The CAN oxidation of N-alkenyl-α-ethoxycarbonylacetamides affords variously functionalised β-lactams through a 4-exo-trig cyclisation of intermediate α-carbamoylalkyl radicals.30
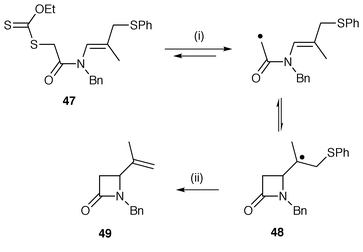 | ||
| Scheme 11 Reagents and conditions: i, dilauroyl peroxide, cyclohexane, reflux; ii, -(PhS·), 48%. | ||
The synthesis of pyrrolidines by cyclisation of radicals, β to the nitrogen atom, onto β-alkenes is now a common protocol. An example of this is the cyclisation of N-tosyl-β-(phenylselanyl)allylamines to yield N-tosylpyrrolidines.31 However, the synthesis of pyrrolidines by cyclisation of radicals, α to the nitrogen atom, onto γ-alkenes is a less common protocol. A novel variant of this protocol uses the reaction between Bu3SnH and N,N-dialkyl-N-(iodomethyl)-but-3-enylamine salts for the synthesis of 5-membered ring ammonium salts.32 The quaternisation improves the Thorpe–Ingold effect and facilitates improved cyclisation in otherwise difficult to cyclise examples.
The addition of a radical generating reagent A–B across two alkenyl groups β to a nitrogen moiety continues to be a common synthetic protocol. The addition of a radical reagent A–B can be R3SnH and is well exemplified in the key step in a synthesis of (−)-α-kainic acid from L-serine using a trimethyltin radical carbocyclisation of a diene precursor33 to make the C3–C4 bond as shown in Scheme 12.34 The trimethyltin radicals are generated in low dilution from trimethyltin hydride, generated by reduction of trimethyltin chloride with sodium cyanoborohydride, and add to the more reactive exo double bond of the diene 50. Subsequent 5-exo cyclisation generates the C3–C4 bond of the pyrrolidine ring generating a new α-methoxycarbonyl stabilised radical which in turn abstracts hydrogen from Me3SnH to yield the synthetic intermediate 51 in good yield. The same cyclisations have also been achieved by using phenylthiol in place of trimethyltin hydride as the radical reagent A–B.35
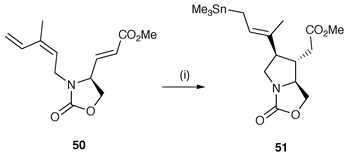 | ||
| Scheme 12 Reagents and conditions: i, Me3SnCl, NaCNBH3, AIBN, t-BuOH, 55% 2.5∶1 trans∶cis. | ||
A novel version of this protocol uses addition of tributyltin radicals onto the aldehyde oxygen atom of α- and β-amino aldehydes 52 to form intermediate O-stannyl ketyl radicals 53 rather than addition onto an alkene (Scheme 13).36 Cyclisation (5- or 6-exo) of the ketyl radicals 53 onto suitably placed double bonds, followed by hydrolytic work-up to hydrolyse off the tributyltin group, gives rise to hydroxy-pyrrolidines 54 and -piperidines with good diastereoselectivity. A novel radical cyclisation used radical addition of toluene-p-sulfonyl bromide as the radical generating reagent A–B across the alkyne bonds of N,N-di(prop-2-ynyl)toluene-p-sulfonamide.37 3,4-Disubstituted pyrrolidines are formed with 80% diastereomeric excess via the addition of TsSePh to dialkyldiallylammonium salts to form the C3–C4 bond of the pyrrolidine ring.38
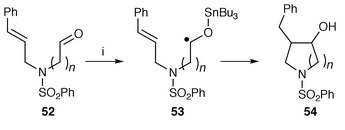 | ||
| Scheme 13 Reagents and conditions: i, Bu3SnH, AIBN, PhH, reflux; n = 1, 52% (exo∶endo = 1.6∶1); n = 2, 56% (dr 1.3∶1). | ||
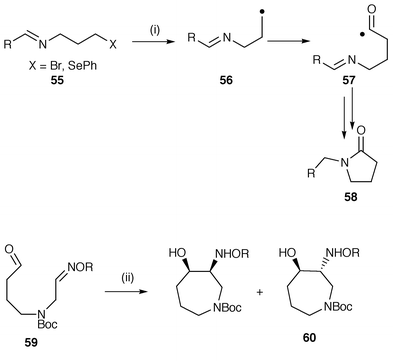
Radical additions to C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) N bonds provides an alternative route to nitrogen heterocycles and two examples are shown in Scheme 14. In the first protocol carbon monoxide is used to trap the intermediate radical.39 The intermediate radical 56 generated from precursor 55 reacts with carbon monoxide under high pressure to generate a new intermediate carbonyl radical 57 which in turn undergoes an apparent 5-exo cyclisation onto the nitrogen of the imine group to yield the pyrrolidin-2-one 58. The intermediate radical 56 is reluctant to undergo 4-exo cyclisation onto the nitrogen of the imine or 5-endo cyclisation onto the imine carbon which allows time for the relatively slow addition of carbon monoxide. The procedure is a 4 + 1 type carbonylation–annulation. In the second protocol the known cyclisation onto oxime ethers is used to generate the hexahydroazepine fragment in the total synthesis of (−)-balanol, a potent inhibitor of protein kinase C.40 The 7-exo cyclisation is unusual but is facilitated by polarity effects wherein cyclisation of the intermediate nucleophilic O-tributyltin ketyl radical onto the electrophilic carbon atom of the oxime ether is strongly favoured. In this reaction the radical generating reagent A–B is Bu3SnH which adds initially to the aldehyde group in the precursor 59. The diastereoselectivity to give preferentially the required trans hexahydroazepine 60 further exhibits the usefulness of this protocol. The use of SmI2 in place of Bu3SnH promoted cyclisation which gave better selectivity in favour of the desired trans isomer. With SmI2 the intermediate is the O-diiodosamarium ketyl.
N bonds provides an alternative route to nitrogen heterocycles and two examples are shown in Scheme 14. In the first protocol carbon monoxide is used to trap the intermediate radical.39 The intermediate radical 56 generated from precursor 55 reacts with carbon monoxide under high pressure to generate a new intermediate carbonyl radical 57 which in turn undergoes an apparent 5-exo cyclisation onto the nitrogen of the imine group to yield the pyrrolidin-2-one 58. The intermediate radical 56 is reluctant to undergo 4-exo cyclisation onto the nitrogen of the imine or 5-endo cyclisation onto the imine carbon which allows time for the relatively slow addition of carbon monoxide. The procedure is a 4 + 1 type carbonylation–annulation. In the second protocol the known cyclisation onto oxime ethers is used to generate the hexahydroazepine fragment in the total synthesis of (−)-balanol, a potent inhibitor of protein kinase C.40 The 7-exo cyclisation is unusual but is facilitated by polarity effects wherein cyclisation of the intermediate nucleophilic O-tributyltin ketyl radical onto the electrophilic carbon atom of the oxime ether is strongly favoured. In this reaction the radical generating reagent A–B is Bu3SnH which adds initially to the aldehyde group in the precursor 59. The diastereoselectivity to give preferentially the required trans hexahydroazepine 60 further exhibits the usefulness of this protocol. The use of SmI2 in place of Bu3SnH promoted cyclisation which gave better selectivity in favour of the desired trans isomer. With SmI2 the intermediate is the O-diiodosamarium ketyl.
 | ||
| Scheme 14 Reagents and conditions: i, CO, AIBN, Bu3SnH, PhH, reflux, 80 atm, 2 h, 70–80%; ii, R = Me, Bu3SnH, AIBN, PhH, reflux, 1∶2, 48%; R = Bn, Bu3SnH, AIBN, refluxing PhH, 1∶2.6, 50%; R = Bn, SmI2, HMPA, t-BuOH–THF, 1∶6.6, 53%. | ||
4 Pyrrolizidines and other bicyclic nitrogen heterocycles
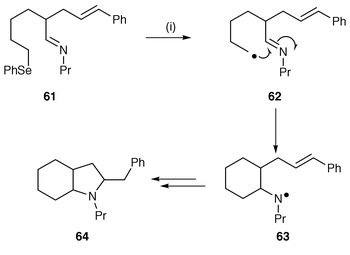
Pyrrolizidines and indolizidines have been common targets for radical cyclisation reactions using a number of different protocols. These protocols have now been applied to a much wider range of different bi- and poly-cyclic nitrogen heterocycles and can be envisaged for synthetic use in most ring systems. We have tried to define both protocols and synthetic targets in this section.The application of synthetic methods using nitrogen centred radicals continue to be used. For instance, in the total synthesis of the indolizidine alkaloid 223AB the final ring closure step was achieved via a Bu3SnH mediated radical cyclisation of a monocyclic chloramine onto a pendant side chain alkene.41 The aminyl radical was generated from the chloroamine using reductive cleavage with CuCl and CuCl2. Iminyl radicals, generated from iminodithiocarbonates [N–N(R)CS2Me] by irradiation with a sun lamp in the presence of small amounts of hexabutylditin, have been used to synthesise several bicyclic nitrogen heterocycles.42 This methodology is similar to that for xanthates shown in Scheme 2. Aminyl radicals generated in tandem reactions, initiated by cyclisation of sp3 carbon-centred radicals onto the electrophilic C-atom of imines, have been used to develop a new protocol for the synthesis of 2-azabicyclo[3.3.0]octanes and perhydroindolines, spirocyclic amines and indolizidines.43 An example of this use of imines as intermediates to aminyl radicals is shown in Scheme 15. Selenides, e.g. 61, are used as the radical precursor to avoid reaction between halide substituted carbon atoms and nucleophilic nitrogen atoms in precursor molecules. Cyclisation of the carbon-centred radical 62 onto the imine generates an intermediate aminyl radical 63 which undergoes 5-exo cyclisation onto the pendant alkene to give the perhydroindoline 64 in good yield. This protocol can be used to synthesise the analogous 5-5, 5-6 and 6-6 nitrogen bicycles. The yields of the reactions in this general protocol are improved by using Lewis acids to increase the electrophilicity of the imine and the intermediate aminyl radicals.
 | ||
| Scheme 15 Reagents and conditions: i, Bu3SnH, AIBN, PhMe, reflux, 57%. | ||
Many of the protocols for synthesising bicyclic nitrogen heterocycles use a nucleophilic nitrogen atom in a larger molecule in alkylation or acylation reactions to attach either a chain containing the group for generating the radical, or a chain containing the unsaturated functionality onto which the radical intermediate will cyclise. Alternatively both chains, i.e. one containing the group for generating the radical and a chain containing the unsaturated functionality onto which the radical intermediate will cyclise, are added to a central nitrogen atom. These alkylation reactions of nitrogen atoms provide fast and facile routes into the syntheses of complex nitrogen heterocyclic targets.
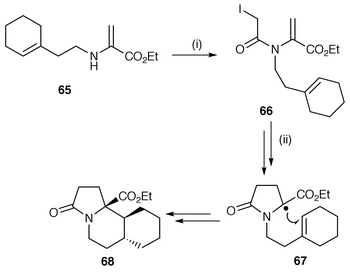
In the first of these protocols, the addition of α-halogenoacyl groups to the nitrogen atom is commonly used. An example of the protocol for the synthesis of pyrrolizidines in which α-iodoacetyl chloride is used in acylation of a suitable nitrogen atom containing precursor is shown in Scheme 16.44 The starting material 65 is acylated, thereby providing a fast route to yield the radical precursor 66. Standard Bu3SnH methodology yields the intermediate stabilised radical 67 by a now well investigated but unusual 5-endo cyclisation. The second cyclisation of intermediate 67 to yield product 68 is an example of a stable radical often preferring to undergo the thermodynamically favoured 6-endo cyclisation rather than the faster 5-exo cyclisation. Other examples in the literature which report the use of this protocol are: α-iodoacetyl chloride for the cyclisation of N-(cycloalk-1-en-1-yl)-α-haloacetamides,45,46
α,α-bis(phenylsulfanyl)acetyl chloride for the synthesis of octahydroindol-2-ones,47 and trichloroacetyl chloride for the synthesis of a range of bicylic heterocycles including tetrahydro-2H-indol-2-ones,48 2-azabicyclo[3.3.1]nonanes,49 and the first total synthesis of the indole alkaloids (±)-melinonine-E and (±)-strychnoxanthine.50 Other variants of this methodology use alkylation with α-bromoacetates to facilitate the synthesis of selenoesters as radical precursors33 and for use with manganese(III) acetate as the radical generating reagent.51 The protocol can be applied the other way round and acylation on nitrogen with acryloyl chloride gives acrylamides which can be used as the alkene moiety onto which radicals can be cyclised to yield γ-lactams.52
 | ||
| Scheme 16 Reagents and conditions: i, ICH2COCl, PhNEt2; ii, Ph3SnH, 5 h, PhH, AIBN, 61%. | ||
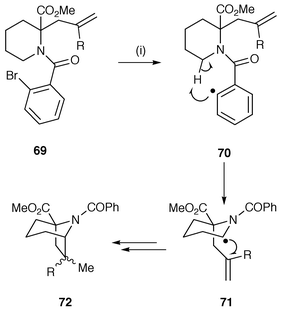
Acylation using ω-halogenoacyl chlorides has also been used in this general protocol, e.g. acylation of 1,4-dihydropyridines using 4-iodobutanoyl chloride has been used to prepare precursors for the syntheses of lupinine and epilupine,53 and 3-bromobutanoyl chloride for the synthesis of the indolizidine framework of pumiliotoxin 251D.54 A different methodology uses acylation with o-bromobenzoyl chloride in order to add a radical generating group which does not itself take part in the cyclisation but instead facilitates 1,5-hydrogen abstraction (Scheme 17).55 Radical abstraction of the bromine from the precursor 69 yields a reactive aryl radical 70 which abstracts a hydrogen atom to yield a new intermediate radical 71 which undergoes 5-exo and 6-endo cyclisation to yield bridged azabicyclic compounds, e.g. 72. A similar protocol uses vinyl radicals instead of aryl radicals for 1,5-hydrogen abstraction for the synthesis of pyrrolizidines.56
 | ||
| Scheme 17 Reagents and conditions: i, Bu3SnH, AIBN, PhMe, reflux, 2 h; R = H, 100% (exo∶endo = 66∶34); R = Me, 40%. | ||
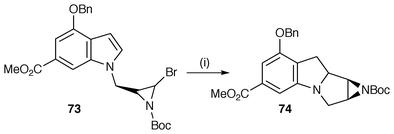
Alkylation using ω-halogenoalkyl halides has also been widely used in this general protocol. A useful illustration of this protocol comes from the ongoing studies of Ziegler and co-workers on the synthesis of mitomycin analogues.57 In this study, an asymmetric route to the core nucleus of the antitumour agent FR-900482 has been facilitated by alkylation with an aziridinylmethyl halide to yield a precursor 73 (Scheme 18). Reaction with Bu3SnH yields an aziridinyl radical which cyclises onto a functionalised indole nucleus to yield the tetracyclic skeleton 74 which is central to this group of antitumour compounds. There is however competition between cyclisation and reduction by Bu3SnH of the intermediate aziridinyl radical to yield the uncyclised aziridine. Alkylation with dibromobutane provides precursors for the radical synthesis of the azepinoindole substructure found in several Stemona alkaloids by an unusual 7-endo radical cyclisation.58 Reductive N-alkylation using 2-(phenylselanyl)ethanal and sodium cyanoborohydride allows the synthesis of 2-(phenylselanyl)ethyl side chains which can be used for generating radicals and cyclising onto suitable alkenes to yield bridged heterocycles, e.g. 1,5-dimethyl-1-azabicyclo[3.2.1]octane iodide.59
 | ||
| Scheme 18 Reagents and conditions: i, Bu3SnH, AIBN, PhMe, 46%. | ||
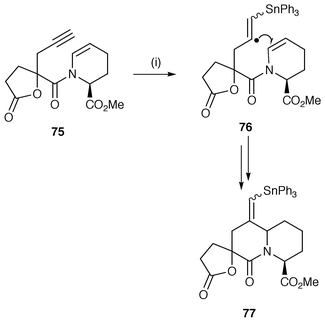
The use of vinyl radicals, resulting from alkylation with ω-(vinyl bromides), has been used in a number of alkaloid synthetic studies, e.g. mossambine,60 vincadifformine61 and the morphinan skeleton.62 Vinyl radicals can also be generated by exo addition of triorganotin hydrides to alkyne precursors, e.g. in the synthesis of (±)-A58365B, an inhibitor of angiostensin converting enzyme (Scheme 19).63 The pendant alkyne is added by acylation of the nitrogen atom to yield the radical precursor 75. The vinyl radical intermediate 76 undergoes 6-exo cyclisation onto the tetrahydropyridine ring to yield a vinyl stannane product 77.
 | ||
| Scheme 19 Reagents and conditions: i, Ph3SnH, AIBN, PhMe, reflux, 62%. | ||
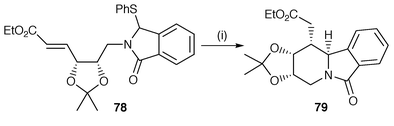
A protocol using alkylation of α-phenylsulfanyl lactams and treatment of the resulting product using Bu3SnH methodology to generate radicals α to the nitrogen has been used for some time. The α-phenylsulfanyl lactam precursors are obtained from the corresponding imide in several simple steps. Several further applications of this methodology have been reported.64,65 In studies aimed towards the synthesis of the indole alkaloid tacamonine, the precursor 78 has been cyclised to yield the all cis tetrahydropyrido[2,1-a]isoindolone 79 stereoselectively as the major diastereomer (Scheme 20).64
 | ||
| Scheme 20 Reagents and conditions: i, Bu3SnH, AIBN, PhH, reflux, 10 h, 60%. | ||
Several papers report the synthesis of bicyclic spiro amines.43,66,67 The methodologies shown in Scheme 15 using tandem cyclisation onto imines43 and the protocol illustrated in Scheme 2 using xanthate transfer66 have also been applied to the synthesis of spiroamines. A general approach to spiro-piperidinyl heterocycles uses aryl radical cyclisation onto alkenes in piperidine rings.67
5 Oxygen heterocycles
Oxygen centred radicals, unlike aminyl or iminyl radicals, are not commonly used in the synthesis of O-heterocycles, but one example of a tandem process featuring an alkoxyl radical cyclisation onto the β-position of an α,β-unsaturated ester has been reported.68 The alcohol is reacted with benzeneselenenyl chloride to generate the sulfenate ester (RO–SePh) radical precursor. Treatment of the selenenate ester with Bu3SnH yields the alkoxyl radical which cyclises to afford 2,3-trans-disubstituted tetrahydrofurans, creating two new contiguous stereogenic centres with high levels of 1,2-induction in each step. The reaction is analogous to the use of sulfenamides for aminyl radical precursors (see Scheme 8).The most commonly used method for the synthesis of O-heterocycles is 5-exo cyclisation of carbon-centred radicals onto suitable unsaturated bonds to yield tetrahydrofuran derivatives. As discussed for the synthesis of nitrogen heterocycles in Section 3 many of the precursors for O-heterocycles are synthesised by addition of the radical generating moiety, or the unsaturated bond moiety, to the oxygen atom of the starting material. This general procedure for the synthesis of tetrahydrofuran derivatives is illustrated in Scheme 21.69 Aryl selenides or aryl tellurides are added to epoxides to yield β-hydroxy-selenides or -tellurides 80 which are alkylated with sodium hydride and allyl bromide to give the radical precursors 81. Standard Bu3SnH methodology gives high yields of tetrahydrofurans 82 whereas the tellurides are sufficiently reactive to undergo radical group transfer using hexabutylditin and light to facilitate the reaction. A number of other examples are reported.70–76
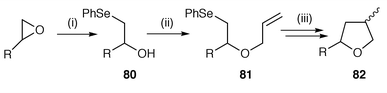 | ||
| Scheme 21 Reagents and conditions: i, ArSe− ; ii, NaH, allyl bromide; iii, Bu3SnH, AIBN. | ||
A novel synthesis and mechanistic study involves radical spirocyclisation to form spirodienones with a tetrahydrofuran ring (Scheme 22).77 The putative biomimetic mechanism outlined in Scheme 22 also helps to explain the biosynthesis of the interiorin and kadsulignan groups of dibenzocyclooctadiene lignans. The Barton ester 83 undergoes homolysis with loss of carbon dioxide to generate an intermediate carbon-centred radical 84 which undergoes 5-exo cyclisation onto the arene to yield the spirodienyl radical 85. An unusual but known rearrangement involving the o-nitro group takes place to yield the spirodienone 86 in good yield.
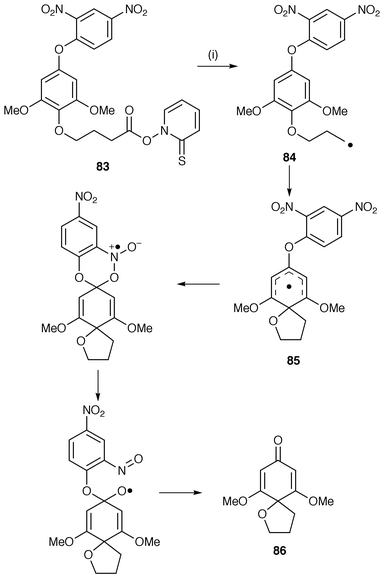 | ||
| Scheme 22 Reagents and conditions: i, hν, -CO2, -(PyrS·). | ||
In the synthesis of tetrahydrofurans a number of radical generating methods have been reported other than the use of Bu3SnH (or related compounds) to abstract a halogen. For instance, O-alkylation with propargyl bromide yields radical precursors for tetrahydrofuran synthesis. Radical addition (phenylthiyl78 and tributylstannyl79) to the alkyne provides an intermediate vinyl radical which rapidly cyclises by 5-exo cyclisation onto a suitable alkene. Other reagents for facilitating radical cyclisation of β-iodo or β-bromo allyl or propargyl ethers to yield a range of polyfunctional tetrahydrofurans include diethylzinc coupled with Pd(II) or Ni(acac)2 as catalyst80 or a catalytic mixed metal system consisting of diethylzinc coupled with MnBr2 and CuCl,81 and a catalytic amount of cobaloxime, regenerated by reduction with sodium borohydride.82 Ring opening of strained three membered rings also provides several protocols for generating carbon-centred radicals for synthesising tetrahydrofurans.83
Six-membered ring cyclic ethers can be synthesised by similar methods to those for tetrahydrofurans. In studies of the synthesis of the potent antitumour agent mucocin A, a novel route uses intramolecular 6-exo radical cyclisation of acyl radicals, generated from acyl selenides.84 The acyl radicals are cyclised onto Z-vinylogous sulfonates to control rotamer population and afford the cis-2,6-disubstituted tetrahydropyran-4-ones in high yields. The protocol has been extended to an iterative process to build up fused polycyclic ethers via acyl radical cyclisations (Scheme 23).85 These fused polycyclic ethers are found in a number of natural products such as the newly discovered gamberic acids. In this iterative process, the first acyl selenide 87 undergoes 6-exo cyclisation via the acyl radical with high cis stereoselectivity to yield the monocyclic ketone 88. The cyclic ketone 88 was converted to the second acyl selenide precursor 89 in a few routine synthetic steps and cyclised again by 6-exo cyclisation with complete stereoselectivity, as required in the natural products, to the bicyclic ether 90. Other protocols for generating six-membered ring cyclic ethers include 6-exo cyclisation of nucleophilic ‘ketyl’ radicals onto the β-position of α,β-unsaturated esters.86,87 The ‘ketyl’ radicals are generated by addition of tributylstannyl radicals86 or samarium diiodide.87
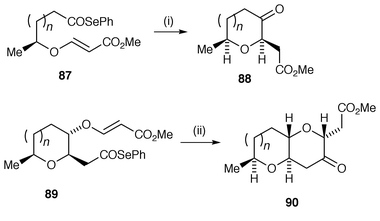 | ||
| Scheme 23 Reagents and conditions: i, (Me3Si)3SiH, Et3B, PhH, rt, air; n = 1, 94%, cis∶trans = 5.7∶1; n = 2, 90%, cis∶trans ≥19∶1; ii, (Me3Si)3SiH, Et3B, PhH, rt, air; n = 1, 91%; n = 2, 99%. | ||
The synthesis of lactones is a common target of radical cyclisation and two of the common protocols are illustrated in Scheme 24. In both of these protocols the radical precursor is synthesised by reaction between the moiety containing the radical generating group and a suitable allylic alcohol group on the starting material. In the first of these protocols α-substituted esters are used as the radical precursors.88,89 In Scheme 24, reaction between the α-iodo ester 91 and Ph3SnH yields the bicyclic lactone 92 in 73% yield.88 The second method uses intermediate β-bromo acetals as radical precursors which are cyclised and subsequently oxidised to yield the lactone.90,91 In the example shown in Scheme 24 the precursor 93 contains a chiral auxiliary.90,91 The cyclisation to the cyclic acetal 94 proceeds with poor diastereoselectivity unless used in tandem with MAD [methylaluminiumbis(2,6-di-tert-butyl-4-methylphenoxide)] when a >98% de was obtained. Hydrolysis and oxidation of the product acetal yields the required target lactone 95. In a third variant of this general protocol, β-bromo- or β-(phenylselanyl)-alcohols have been used to form esters as precursors for radical cyclisation to γ- and δ-lactones for the synthesis of α-hydrazino- and α-(O-benzylhydroxylamino)-lactones in 70–82% yields.92 Alternative methods for the synthesis of lactones use Mn(OAc)3 mediated oxidation of α-keto esters onto suitable alkenyl side chains93 and SmI2 mediated radical addition of ketones onto α,β-unsaturated esters followed by cyclisation.94
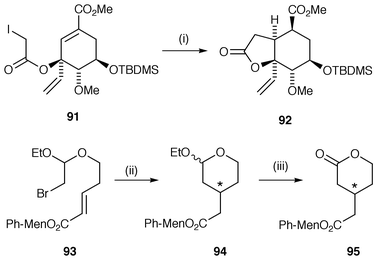 | ||
| Scheme 24 Reagents and conditions: i, Ph3SnH, AIBN, PhH, reflux; ii, Bu3SnH, Et3B, MAD, toluene, −40 °C, 1.5 h, 38%, de > 98%; iii, 10% HClO4, Ag2CO3. | ||
Radical cyclisation to yield cyclic peroxides has proved useful for the synthesis of analogues of the important antimalarial compound yingzhaosu isolated from a Chinese herbal medicine which has the unusual 2,3-dioxabicyclo[3.3.1]nonane skeleton.95 A very novel radical cascade reaction which includes molecular oxygen in the mechanism has been used to synthesise this cyclic peroxide 98 which is illustrated in Scheme 25. In the synthesis, (−)-carvone 96 is used as the precursor for the radical addition of oxygen and also contains the required chiral centre which determines the stereochemistry of the final product. The sequence is initiated by addition of phenylthiyl radicals to the exo alkene to yield a radical intermediate which is trapped by oxygen to yield the peroxyl radical 97. 6-exo Cyclisation of the peroxyl radical 97 gives the required 2,3-dioxabicyclo[3.3.1]nonane skeleton and finally the cyclic peroxide product 98. The synthesis of 1,2,4-trioxanes and 1,2,4-trioxepanes by NBS/NIS mediated radical cyclisations of unsaturated hydroperoxyacetals has also been reported.96
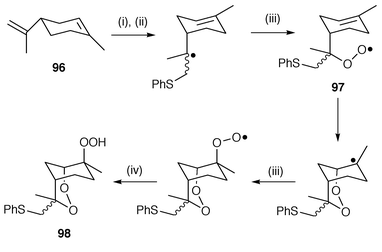 | ||
| Scheme 25 Reagents and conditions: i, PhSH, O2, DBPO; ii, PhS·; iii, O2; iv, PhSH→PhS·. | ||
6 Carbohydrates and nucleosides
Radical cyclisation has been used in the synthesis of novel carbohydrates as well as using carbohydrates as templates for radical cyclisation.97,98 One key advantage of the use of radical cyclisation with carbohydrates is that the hydroxy groups do not normally need protection which can save steps in a synthesis. An example of the use of carbohydrate templates in synthesis is shown in Scheme 26 in which the functionalised monosaccharide 99 is converted to the functionalised cyclopentane derivative 100.97 This synthetic sequence illustrates the application of the Stork protocol using cyclisation of silyl methylene radicals for the introduction of new hydroxy functions. An example of the use of radical cyclisation in the synthesis of an optically pure C-glycoside 102 is shown in Scheme 27.99 Bu3SnH or PET mediated 5-exo cyclisation of the tartaric acid derivative 101via ketyl radical intermediates gave reasonable yields of 102 with high stereoselectivity.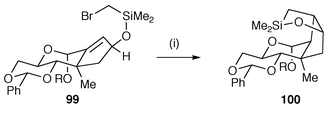 | ||
| Scheme 26 Reagents and conditions: i, R = Me, Bu3SnCl, NaCNBH3, AIBN, t-BuOH, 15 h, 90 °C. | ||
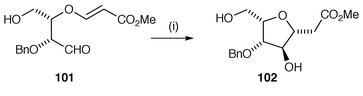 | ||
| Scheme 27 Reagents and conditions: i, Bu3SnH, AIBN, 40%. | ||
7 Sulfur, selenium and tellurium heterocycles
Free radical chemistry involving sulfur, selenium and tellurium has some marked differences to that of oxygen and nitrogen because of their soft or polarisable nature and the longer and weaker C–S, C–Se, C–Te bonds. SH2 reactions centred on S, Se and Te have become important and have been used to advantage by Schiesser and co-workers.100–102 Two of the synthetic reactions using these mechanistic possibilities are illustrated in Scheme 28. In the first the (phenyltelluro)formate precursor 103 undergoes homolysis facilitated by light catalysis to yield intermediate oxyacyl radical 104. Intramolecular SH2 (SHi) substitution by the acyl radical on selenium with benzyl radical as the leaving group yield the cyclic selenium heterocycle 105 in good yield. The protocol works best with n = 2 and when n > 2 other mechanisms predominate and yields decline. Reaction of 2-methyl-2-(2-iodophenyl)oxirane 106 with two equivalents of sodium butyltelluroate (NaTeBu), generated from sodium borohydride reduction of dibutyl ditelluride in THF, affords 2,3-dihydro-3-hydroxy-3-methyl-1-benzotellurophene 108via the hydroxy telluride 107 in 57% yield. This mechanism has been shown to involve a tandem SRN1–SHi sequence. In a further example, γ-thiolactones have been synthesised by carbonylation with carbon monoxide of intermediate free radicals to yield acyl radicals which undergo cyclisation by intramolecular SH2 (SHi) at sulfur.103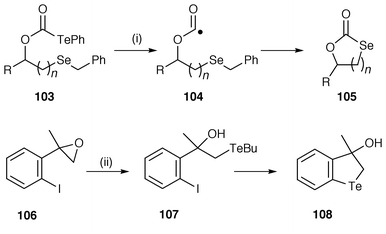 | ||
| Scheme 28 Reagents and conditions: hν, n = 2, 73%; ii, BuTe, THF, 57%. | ||
Motherwell and co-workers have carried out several syntheses of unusual S-heterocycles involving cyclisation of aryl radicals onto side chains containing sulfonates and sulfonamides.104–106 In Scheme 29 the synthesis of a biphenyl system containing a seven membered ring sulfonamide is shown as an illustration of this methodology.104 The benzylic N-methylsulfonamide 109 is converted to the cyclised sulfonamides 110 and 111. In this ipso substitution onto arenes there is normally an oxidative step leading to the rearomatised product 110. These oxidative steps in Bu3SnH reactions are now well known if there is a driving force to rearomatisation. In some of these reactions the dihydro products without rearomatisation, e.g. 111, are isolable. In this example, benzylic sulfonates and their corresponding benzylic N-methylsulfonamides prefer to undergo [1,7] addition reactions rather than [1,6] ipso-substitution. ortho-Substituents effectively counteract this tendency for addition. The guidelines104,105 for this synthetic strategy of ipso substitution indicate that the synthesis of biphenyls is favoured by the location of electron releasing o-substituents around the sulfonyl substituted acceptor ring, thereby leading to hindered products. 4-Aryl-5,6-dihydro-1,2-oxathiine 2,2-dioxides and related heterocyclic systems have been synthesised using this protocol which also involves an unusual rearrangement reaction via radical addition to aryl and heteroaryl rings.106
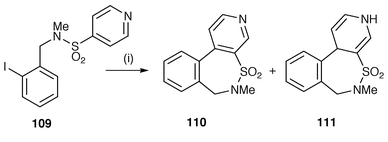 | ||
| Scheme 29 Reagents and conditions: i, Bu3SnH, AIBN, slow addition, PhH, reflux; 110 (55%); 111 (24%). | ||
The other characteristic of radical reactions involving sulfur atoms which is uncommon for those involving nitrogen or oxygen atoms is the ability to undergo β-scission to give intermediate thiyl radicals. A number of examples in the literature have shown the synthetic potential of using this behaviour for the preparation of thiophenes,107 dihydrothiophenes108 and 2,3-dihydrobenzothiophenes.109
8 Silicon heterocycles
The syntheses of silyloxy heterocycles (with Si–O in the ring) are primarily of interest as synthetic methods rather than as an end in themselves. There has been growing interest in the use of silicon containing tethers attached to hydroxy groups in radical cyclisation for the introduction of new C–C bonds.4 The well known Stork protocol uses cyclisation of silyl methylene radicals onto alkenes for the introduction of new hydroxy functions. The silyloxy group is oxidised out of the ring to leave two hydroxy groups. An example of the use of this radical cyclisation in synthesis is shown in Scheme 26.97 New examples use 5-exo-trig cyclisations of radicals generated from (bromomethyl)dimethylsilyl propargyl ethers for the synthesis of 5-membered rings with silicon and oxygen heteroatoms,110 and from (bromomethyl)dimethylsilyl esters of α,β-unsaturated carboxylic acids, for the synthesis of silalactones.111 A number of examples use cyclisation onto the silicon containing tethers which also have attached alkenes112 or alkynes.1139 Benzoheterocycles
The synthesis of benzoheterocycles using radical cyclisation can be envisaged by several general routes: a, cyclisation of aryl radicals onto side chain unsaturated bonds, b, cyclisation of aryl radicals onto pendant arenes/heteroarenes and c, cyclisation of side chain radicals onto arenes/heteroarenes.In Scheme 30 the use of cyclisations of radicals generated from N-substituted 7-bromoindoles for the synthesis of tricyclic indole analogues is illustrated.114 The butenyl derivative 112 undergoes 6-exo cyclisation to yield the tricyclic indole 113. The analogous N-allyl-7-bromoindole undergoes 6-endo rather than 5-exo cyclisation due to constraints enforced on the system by the geometry of the indole ring. N-(ω-Alkenyl)-2-bromoindoles react to yield intermediate indol-2-yl radicals which undergo 5-exo or 6-exo cyclisation onto the pendant alkene to yield [1,2-a]-fused pyrroles. Indolines have been prepared by cyclisation of aryl radicals generated from o-halogeno N-alkenylanilines.115 Other Bu3SnH mediated aryl radical cyclisations have been used to synthesise isoindolones from N-ethenyl-2-bromobenzamides leading to the synthesis of alkaloids lennoxamine and chilenine,116 isoquinolinones from 2-(2-bromophenyl)-N-(ethenyl)phenylacetamides,117 2,3-dihydrobenzothiophenes,118 2,3-dihydrobenzofurans119 and isoquinolones and quinazolones.120 Cyclisation of aryl radicals onto side chain unsaturated bonds using protocols other than Bu3SnH include: 2,3-dihydrobenzofurans using PhMgBr–FeCl2,121 carbon monoxide with tri(perfluoroalkyl)tin hydrides122 and lithium tributylmanganate (n-Bu3MnLi).122 Indolines have also been synthesised using lithium tributylmanganate.123
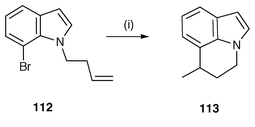 | ||
| Scheme 30 Reagents and conditions: i, Bu3SnH, AIBN, PhMe, reflux, 12 h. | ||
Cyclisation of aryl radicals onto pendant arenes provides a useful method of constructing biphenyl moieties within polycyclic heterocycles. Phenanthridones have been usefully synthesised by this protocol (Scheme 31).124 The o-iodobenzamide 114 forms an aryl radical 115 which can cyclise by a 5-exo route to 116 followed by a neophyl rearrangement to 117 or by a 6-endo route directly to intermediate radical 117. This unknown aspect of the mechanism of these reactions has been solved by using trapping with benzeneselanol (PhSeH) which is an extremely fast hydrogen donor; radical 116 was trapped with PhSeH to yield the spirocyclohexadiene 118 to provide strong evidence for 5-exo cyclisation. The intermediate radical 117 rearomatises to yield the phenanthridone 119 by an unknown mechanism which is usual for this synthetic sequence, i.e. an oxidation step in a Bu3SnH mediated cyclisation. This general protocol has been used for the synthesis of phenanthridines leading to the syntheses of the Amaryllidaceae alkaloids, vasconine, assoanine, oxoassinine and pratosine.125 The protocol is attractive by its simplicity; o-bromobenzyl bromides are reacted with anilines to yield the radical precursors. The mechanism of the oxidative step is unclear and although a large amount of AIBN is required it does not appear to act as the oxidant.125 This protocol has also been reported for converting (o-bromobenzyl)phenyl ethers to 6H-dibenzo[b,d]pyrans and a range of precursors with various methoxy and methylenedioxy substituents have been used with success.126
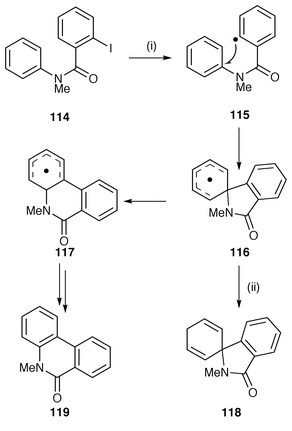 | ||
| Scheme 31 Reagents and conditions: i, Bu3SnH, AIBN, PhMe, reflux; ii, PhSeH. | ||
This methodology has also been applied to cyclisation of aryl radicals onto heteroarenes (Scheme 32).127 Radical cyclisation of the N-(2′-iodophenyl)pyrrole-3-carboxamide 120 gave the tricyclic pyrrolo[3,2-c]quinolone 121, the ring system found in the recently isolated alkaloid martinelline. This oxidative Bu3SnH mediated cyclisation of aryl radicals has been used to convert N-aroyl-6-bromoindoles into the pyrrolophenanthridone alkaloids, hippadine, pratosinine, pratorimine and pratorinine from various Crinum species (Amaryllidaceae).128 The protocol has also been used for the preparation of the pyrrolophenanthridine alkaloids oxoassoanine and anhydrolycorin-7-one from N-aroyl-6-bromoindolines.129
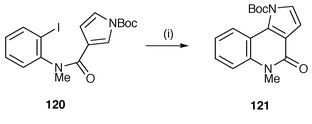 | ||
| Scheme 32 Reagents and conditions: i, Bu3SnH, AIBN, PhMe, reflux, 52%. | ||
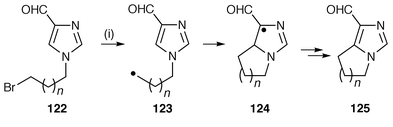
Cyclisation of N-(ω-alkyl) radicals onto heteroarenes has been used to annulate pyrrole, imidazole130 and indole.131 This oxidative radical cyclisation using Bu3SnH is illustrated in Scheme 33 for the synthesis of [1,2-c]-fused imidazoles 125 from imidazolecarbaldehydes 122.130 [1,2-a]-Fused pyrroles have been prepared by the same protocol from acylpyrroles. The intermediate nucleophilic N-alkyl radicals 123, generated from the precursor 122, cyclise onto the imidazole ring to yield the π-radical 124 which undergoes oxidative rearomatisation to 125. This methodology was applied successfully to 5-, 6-, and 7-membered rings but fails with larger ring cyclisations. A chain mechanism involving single electron transfer has been proposed to explain the unusual oxidative step observed in all these reactions.
 | ||
| Scheme 33 Reagents and conditions: i, Bu3SnH, AIBN, MeCN, reflux, 5 h; n = 1, 42%; n = 2, 49%; n = 3, 14%. | ||
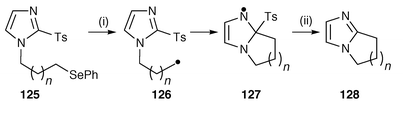
Regioselective (ipso) aromatic homolytic substitution has been applied to the syntheses of [1,2-a]-fused-benzimidazoles,132 -imidazoles,132 -indoles133,134 and uracils.134 Treatment of imidazole precursors 125 with Bu3SnH under standard radical conditions gave the cyclisation products 128 in reasonable yields without the formation of any uncyclised reduced products (Scheme 34). In these reactions the initial radical 126 cyclises onto the electrophilic C-2 position to yield a π-radical intermediate 127 which rearomatises with loss of tosyl radicals to yield the [1,2-a]-fused imidazoles 128. The tosyl radicals react with Bu3SnH to regenerate tributylstannyl radicals for carrying the chain reaction.
 | ||
| Scheme 34 Reagents and conditions: i, Bu3SnH, AIBN, PhMe, reflux; n = 1, 52%; n = 2, 48%; n = 3, 63%. | ||
10 Heteroarenes
The synthesis of heteroarenes by radical cyclisation is a surprisingly under-studied area with few general protocols. One of the few novel methodologies is illustrated in Scheme 1 for the synthesis of camptothecin. Another novel methodology, by Murphy and co-workers,135 uses diazonium salts as precursors for the synthesis of indoles which is exemplified in Scheme 35. Reaction between the precursor diazonium salts 129 and iodide yields the initial aryl radical intermediates 130 which undergo 5-exo cyclisation onto the vinyl bromides followed by β-elimination of bromide to yield 131. The exo-alkene shifts into the 2,3-position to yield the indoles 132. This new route avoids the need to use trialkyltin reagents. 2-Cyanoaryldiazonium salts have also been used in a novel cascade radical reaction to generate 2-cyanoaryl radicals which add to aryl isothiocyanates to give α-(arylthio)imidoyl radicals which undergo a 5-exo-dig cyclisation onto the cyano group eventually leading to tetra condensed nitrogen heterocycles.136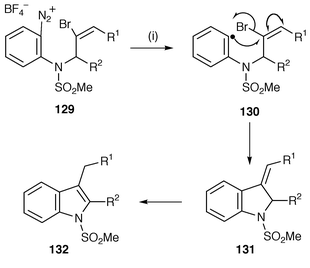 | ||
| Scheme 35 Reagents and conditions: i, NaI, acetone; R1 = Ph, R2 = Me, 49%; R1 = Me, R2 = H, 83%. | ||
Flash vacuum pyrolysis (FVP) of 2-allyloxypropenoic esters give 1-benzofurans,137 and oxime ethers give carbazoles, phenanthridines and acridines.138 Thermal decomposition of tert-butyl o-(phenoxy)- and o-(anilino)-phenyliminoxyperacetates yield acridine, quinazolinone and indole derivatives.139 2-Cyanophenylthiyl radicals, generated by photolysis of bis(2-cyanophenyl) disulfide, add to aryl isonitriles to yield imidoyl radicals which cyclise onto the cyano group to eventually afford thienoquinoxalines.140
11 Macrocyclisation
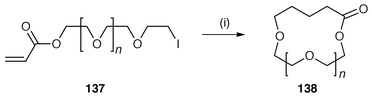
The formation of rings with more than seven atoms have unfavourable rates and in radical cyclisation the interception of intermediate radicals by hydrogen donors such as Bu3SnH is a problem. The most common method of overcoming this rate problem is to use favourable polarity, e.g. cyclise nucleophilic alkyl radical onto an electrophilic centre, commonly the β-position of an α,β-unsaturated ketone, ester or amide. Pattenden and co-workers5,141,142 have studied this methodology extensively and an example of macrocyclisation and transannulation is shown in Scheme 36. The initial radical 134 generated from the acyclic precursor 133 undergoes 12-membered ring endo macrocyclisation to intermediate radical 135 which in turn undergoes transannulation to ultimately yield the bicyclic lactone 136 with stereoselective formation of a trans ring junction.141 An excellent example of the polarity effect of the α,β-unsaturated ester is in the exclusive endo cyclisation of ω-iodo-poly(oxaalkyl) acrylates 137 to the corresponding cyclic polyethers 138 in excellent yields (Scheme 37).143 Interestingly, these poly(oxaalkyl) acrylates undergo radical cyclisations more readily than carbon analogues.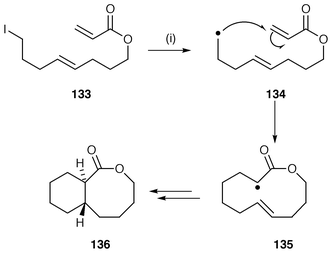 | ||
| Scheme 36 Reagents and conditions: i, Bu3SnH, AIBN, PhH, reflux, 61%. | ||
 | ||
| Scheme 37 Reagents and conditions: i, Bu3SnH, AIBN, PhH, reflux; n = 1, 76%; n = 2, 72%; n = 3, 70%; n = 4, 63%; n = 5, 30%. | ||
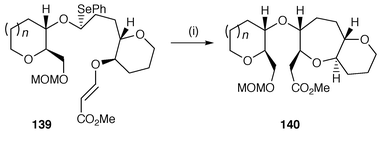
Cyclisation to give 8–10-membered rings requires the same methodology as the larger rings. Radical cyclisation of aryl radicals onto the β-position of α,β-unsaturated amino esters gives good yields of 7–9-membered ring benzocyclic α-amino esters which have the nitrogen atom in the ring.144 Cyclisation to 10-membered rings was unsuccessful.144 In studies towards the synthesis of polycyclic ethers, 7-membered ring cyclisation onto the β-position of β-alkoxy acrylates 139 yields 2,7-disubstituted oxepanes 140 with selective cis stereochemistry at the 2- and 7-positions of the oxepane.145 An example of these cyclisations is shown in Scheme 38. The advantage of polarisation can be used in the opposite sense as well, i.e. a strongly electrophilic radical can be cyclised onto an electron rich alkene. An example of an 8-endo radical cyclisation to yield an eight-membered ring lactone uses cyclisation of a carbon centred radical α to an ester (oxycarbonylmethyl radical).146 The cyclisation of aryl radicals onto constrained side chain alkenes gives selective 8-endo cyclisation. Furo[3,2-c][2]benzoxocines147 and 3,4,5,6-tetrahydro-1-benzazocin-2(1H)-ones148 have been synthesised using this 8-endo cyclisation protocol.
 | ||
| Scheme 38 Reagents and conditions: i, Bu3SnH, AIBN, Et3B, PhH, reflux; n = 1, 82%; n = 2, 87%. | ||
12 Reagents for radical cyclisation
The synthesis of heterocycles using radical cyclisation depends on the same advances in radical synthetic methodology as non-heterocyclic systems. The use of Bu3SnH continues to be dominant and a very useful reagent. The purification problems can be partially overcome by using Bu3SnH catalytically. In this methodology Bu3SnCl is reduced in situ with sodium cyanoborohydride or sodium borohydride thereby producing small amounts of Bu3SnH which is continually used up generating more Bu3SnX (X = Br, I) in a cycle.34,69 This also has the advantage of keeping the concentration of Bu3SnH low to facilitate cyclisation instead of reduction. An example is the reduction and cyclisation of bromoketals in a tandem reaction.69 Tributylgermanium hydride (Bu3GeH) is much less toxic and reacts slightly slower thereby facilitating improved cyclisation yields over use of Bu3SnH but is very expensive. An example is the use in the cyclisation of perfluoroalkenyl radicals.73 Tris(trimethylsilyl)silane [Me3Si)3SiH] is also less toxic and easier to work-up than Bu3SnH but is also expensive.33,49,50 While AIBN [azobis(isobutyronitrile)] is the most commonly used radical initiator, there are other diazene initiators, e.g. AMBN [azobis(methylisobutyronitrile) = Et(Me)C(CN)–N N–C(CN)(Me)Et] which is more soluble and can be used in cyclohexane as well as in toluene as solvent.17 The use of triethylborane and oxygen as an initiator is useful because it allows reactions to be carried out at low or room temperature.25,46,48,112 Triethylborane can also be used to initiate iodine atom transfer reactions. Cyclohexane is becoming the preferred solvent for Bu3SnH mediated reactions because of evidence of the possibility of toluene and benzene participating in radical reactions rather than acting only as a solvent. It is also lower boiling than toluene.17,42One major advance in facilitating environmentally acceptable radical reagents has been the use of fluoroalkyl(fluorous)tin hydride.149 An example of the reagents developed by Curran and co-workers is (C6F13CH2CH2)3SnH. The insolubility of the fluorous reagents in normal organic solvents and water has allowed easy separation of the tin reagents and products. A further development on the use of fluorous tin hydride has been the use of supercritical CO2. This development is in the very early stages but eventually should serve as the reaction and separation solvent. At present, solubility problems need to be overcome but the potential for clean heterocycle synthesis has been exemplified by the synthesis of indolines in high yield.
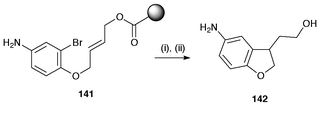
Another important advance is the use of solid phase organic synthesis in radical reactions.150,151 The major advantage with solid phase synthesis is that the radical precursor is attached to the resin and the Bu3SnH used in the reaction can be washed off when the radical cyclisation is complete, thereby eliminating purification problems and lowering problems of toxicity. An example of the use of solid phase synthesis for 2,3-dihydrobenzofurans is shown in Scheme 39. In this preparation the precursor 141 is attached to TentaGel resin beads and after radical cyclisation the product heterocycle 142 is removed by standard methods. Solid phase synthesis has also been used for the radical synthesis of 2,3-dihydrobenzofurans using samarium diiodide.151
 | ||
| Scheme 39 Reagents and conditions: i, Bu3SnH, AIBN, PhMe–t-BuOH; ii, NaOMe–MeOH, rt, >90%. | ||
A number of alternative reagents to triorganotin hydride continue to be used, e.g. use of nickel complex catalysed electroreduction,152 triethylamine mediated photoinduced electron transfer reactions (PET), 153 catalytic cobaloxime regenerated by reduction with sodium borohydride,81 manganese triacetate,51,154 lithium tributylmanganate (n-Bu3MnLi)123 and samarium diiodide.73,87,151
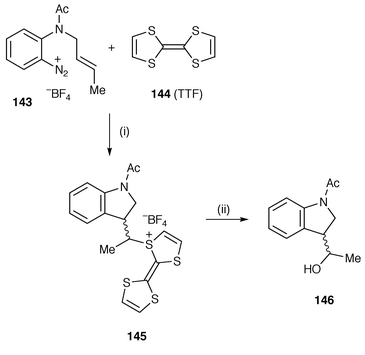
Murphy and co-workers have continued to develop their novel use of tetrathiafulvalene 144 (TTF) for the reduction of aryldiazonium salts as a method of generating aryl radicals.155 TTF is an excellent electron donor and transfers an electron to the diazonium salt to yield intermediate aryldiazene radicals (ArNN·) which are unstable and rapidly lose nitrogen to yield aryl radicals. Aryldiazonium salts are readily accessed from anilines and are cleanly reduced to aryl radicals. An example of the synthesis of indoline 146 from diazonium salt 143 is shown in Scheme 40. The intermediate cyclised radical is trapped by the TTF to yield the intermediate TTF adduct 145 which undergoes hydrolysis to 146. This method allows anilines to be used as precursors and avoids the use of the toxic and troublesome Bu3SnH for generating aryl radicals.
 | ||
| Scheme 40 Reagents and conditions: i, Acetone (moist), 59%; ii, water. | ||
Zard and co-workers1,10,11,27,42,66 have developed their xanthate methodology as a promising alternative to the use of Bu3SnH and have applied it to a wide range of heterocyclic syntheses, including β-lactams,27 spirocyclic amines43 and the tetracyclic alkaloid (±)-matrine.10 The overall protocol is shown in Scheme 2 and another example for the synthesis of β-lactams27 in Scheme 11.
References
- S. Z. Zard, Angew. Chem., Int. Ed. Engl., 1997, 36, 672 CrossRef; B. Quiclet-Sire and S. Z. Zard, Pure Appl. Chem., 1997, 69, 645 CAS.
- V. Nair, J. Mathew and J. Prabhakaran, Chem. Soc. Rev., 1997, 127 RSC.
- S. Kim, Pure Appl. Chem., 1996, 68, 623 CAS.
- L. Fensterbank, M. Malacria and S. McN. Sieburth, Synthesis, 1997, 813 CrossRef CAS.
- S. Handa and G. Pattenden, Contemp. Org. Synth., 1997, 4, 196 RSC.
- E. Lee, Pure Appl. Chem., 1996, 68, 631 CAS.
- D. P. Curran, H. Liu, H. Josien and S.-B. Ko, Tetrahedron, 1996, 52, 11385 CrossRef CAS.
- H. Josien, S.-B. Ko, D. Bom and D. P. Curran, Chem. Eur. J., 1998, 4, 67 CrossRef CAS.
- H. Josien and D. P. Curran, Tetrahedron, 1997, 53, 8881 CrossRef CAS.
- R. N. Saicic and S. Z. Zard, Chem. Commum., 1996, 1631 RSC.
- L. Boiteau, J. Boivin, A. Liard, B. Quiclet-Sire and S. Z. Zard, Angew. Chem., Int. Ed., 1998, 37, 1128 CrossRef CAS.
- J. H. Rigby and M. E. Mateo, Tetrahedron, 1996, 52, 10569 CrossRef CAS.
- H. Takayama, F. Watanabe, M. Kitajima and N. Aimi, Tetrahedron Lett., 1997, 38, 5307 CrossRef CAS.
- S. Atarashi, J.-K. Choi, D.-C. Ha, D. J. Hart, D. Kuzmich, C.-S. Lee, S. Ramesh and S. C. Wu, J. Am. Chem. Soc., 1997, 119, 6226 CrossRef CAS.
- M. D. Bachi and A. Melman, Pure Appl. Chem., 1998, 70, 259; M. D. Bachi and A. Melman, J. Org. Chem., 1997, 62, 1896 CrossRef CAS; M. D. Bachi, N. Bar-Ner and A. Melman, J. Org. Chem., 1996, 61, 7116 CrossRef CAS.
- E. Lee, J. W. Lim, C. H. Yoon, Y.-S. Sung and Y. K. Kim, J. Am. Chem. Soc., 1997, 119, 8391 CrossRef CAS.
- W. R. Bowman, M. J. Broadhurst, D. R. Coghlan and K. A. Lewis, Tetrahedron Lett., 1997, 38, 6301 CrossRef CAS.
- A. J. Clark and J. L. Peacock, Tetrahedron Lett., 1998, 39, 1265 CrossRef CAS.
- O. M. Musa, J. H. Horner, H. Shahin and M. Newcomb, J. Am. Chem. Soc., 1996, 118, 3862 CrossRef CAS.
- M. Newcomb, O. M. Musa, F. N. Martinez and J. H. Horner, J. Am. Chem. Soc., 1997, 119, 4569 CrossRef CAS.
- B. J. Maxwell and J. Tsanaktsidis, J. Am. Chem. Soc., 1996, 118, 4276 CrossRef CAS.
- C. Ha, O. M. Musa, F. N. Martinez and M. Newcomb, J. Org. Chem., 1997, 62, 2704 CrossRef CAS.
- J. H. Horner, O. M. Musa, A. Bouvier and M. Newcomb, J. Am. Chem. Soc., 1998, 120, 7738 CrossRef CAS.
- M.-H. Le Tadic-Biadatti, A.-C. Callier-Dublanchet, J. H. Horner, B. Quiclet-Sire, S. Z. Zard and M. Newcomb, J. Org. Chem., 1997, 62, 559 CrossRef CAS.
- M. Ikeda, H. Teranishi, K. Nozaki and H. Ishibashi, J. Chem. Soc., Perkin Trans. 1, 1998, 1691 RSC.
- K. Goodall and A. F. Parsons, Tetrahedron Lett., 1997, 38, 491 CrossRef CAS.
- L. Boiteau, J. Boivin, B. Quiclet-Sire, J.-B. Saunier and S. Z. Zard, Tetrahedron, 1998, 54, 2087 CrossRef CAS.
- J. Cassayre, B. Quiclet-Sire, J.-B. Saunier and S. Z. Zard, Tetrahedron, 1998, 54, 1029 CrossRef CAS.
- M. Ikeda, S. Ohtani, T. Yamamoto, T. Sato and H. Ishibashi, J. Chem. Soc., Perkin Trans. 1, 1998, 1763 RSC; H. Ishibashi, C. Kameoka, K. Kodama, H. Kawanami, M. Hamada and M. Ikeda, Tetrahedron, 1997, 53, 9611 CrossRef CAS; H. Ishibashi, K. Kodama, C. Kameoka, H. Kawanami and M. Ikeda, Tetrahedron, 1996, 52, 13867 CrossRef CAS.
- A. D’Annibale, A. Pesce, S. Resta and C. Trogolo, Tetrahedron Lett., 1997, 38, 1829 CrossRef CAS.
- V. Gupta, M. Besev and L. Engman, Tetrahedron Lett., 1998, 39, 2429 CrossRef CAS.
- E. W. Della, A. M. Knill and P. A. Smith, Chem. Commun., 1996, 1637 RSC.
- J. Quirante, C. Escolano and J. Bonjoch, Synlett, 1997, 179 CAS.
- S. Hanessian and S. Ninkovic, J. Org. Chem., 1996, 61, 5418 CrossRef CAS.
- O. Miyata, Y. Ozawa, I. Ninomiya, K. Aoe, H. Hiramatsu and T. Naito, Heterocycles, 1997, 46, 321 Search PubMed.
- A. F. Parsons and R. M. Pettifer, Tetrahedron Lett., 1997, 38, 5907 CrossRef CAS; A. F. Parsons and R. M. Pettifer, J. Chem. Soc., Perkin Trans. 1, 1998, 651 RSC.
- S. Caddick, C. L. Shering and S. N. Wadman, Chem. Commun., 1997, 171 RSC.
- M. P. Bertrand, S. Gastaldi and R. Nouguier, Synlett, 1997, 1420 CAS.
- I. Ryu, K. Matsu, S. Minakata and M. Komatsu, J. Am. Chem. Soc., 1998, 120, 5838 CrossRef CAS.
- M. Miyabe, M. Torieda, K. Inoue, K. Tajiri, T. Kiguchi and T. Naito, J. Org. Chem., 1998, 63, 4397 CrossRef CAS.
- T. Momose, M. Toshima, S. Seki, Y. Koike, N. Toyooka and Y. Hirai, J. Chem. Soc., Perkin Trans. 1, 1997, 1315 RSC.
- A.-C. Callier-Dublanchet, B. Quiclet-Sire and S. Z. Zard, Tetrahedron Lett., 1997, 38, 2463 CrossRef CAS.
- W. R. Bowman, P. T. Stephenson and A. R. Young, Tetrahedron, 1996, 52, 11445 CrossRef CAS.
- S. R. Baker, A. F. Parsons, J.-F. Pons and M. Wilson, Tetrahedron Lett., 1998, 39, 7197 CrossRef CAS.
- H. Ishibashi, Y. Fuke, T. Yamashita and M. Ikeda, Tetrahedron: Asymmetry, 1996, 7, 2531 CrossRef CAS.
- M. Ikeda, H. Teranishi, N. Iwamura and H. Ishibashi, Heterocycles, 1997, 45, 863 Search PubMed.
- M. Ikeda, S. Ohtani, M. Okada, E. Minakuchi, T. Sato and H. Ishibashi, Heterocycles, 1998, 47, 181 Search PubMed.
- H. Ishibashi, M. Higuchi, M. Ohba and M. Ikeda, Tetrahedron Lett., 1998, 39, 75 CrossRef CAS.
- J. Quirante, C. Escolano, L. Costejà and J. Bonjoch, Tetrahedron Lett., 1997, 38, 6901 CrossRef CAS; J. Quirante, C. Escolano, M. Massot and J. Bonjoch, Tetrahedron, 1997, 53, 1391 CrossRef CAS.
- J. Quirante, C. Escolano, A. Merino and J. Banjoch, J. Org. Chem., 1998, 63, 968 CrossRef CAS.
- D. T. Davies, N. Kapur and A. F. Parsons, Tetrahedron Lett., 1998, 39, 4397 CrossRef CAS.
- C. Andrés, J. P. Duque-Soladana, J. P. Iglesias and R. Pedrosa, Tetrahedron Lett., 1996, 37, 9085 CrossRef CAS.
- P. Mangeney, L. Hamon, S. Rausson, N. Urbain and A. Alexakis, Tetrahedron, 1998, 54, 10349 CrossRef CAS.
- J. Cossy, M. Cases and D. G. Pardo, Bull. Soc. Chim. Fr., 1997, 134, 141 Search PubMed.
- M. Ikeda, Y. Kugo and T. Sato, J. Chem. Soc., Perkin Trans. 1, 1996, 1819 RSC; M. Ikeda, Y. Kugo, Y. Kondo, T. Yamazaki and T. Sato, J. Chem. Soc., Perkin Trans. 1, 1997, 3339 RSC.
- J. Robertson, M. A. Peplow and J. Pillai, Tetrahedron Lett., 1996, 37, 5825 CrossRef CAS.
- F. E. Ziegler and M. Belema, J. Org. Chem., 1997, 62, 1083 CrossRef CAS.
- J. H. Rigby, S. Laurent, A. Cavezza and M. J. Heeg, J. Org, Chem., 1998, 63, 5587 CrossRef CAS.
- E. W. Della and A. M. Knill, Tetrahedron Lett., 1996, 37, 5805 CrossRef CAS; E. W. Della and A. M. Knill, J. Org. Chem., 1996, 61, 7529 CrossRef CAS.
- M. E. Kuehne, T. Wang and D. Seraphin, J. Org. Chem., 1996, 61, 7873 CrossRef CAS.
- M. E. Kuehne, T. Wang and P. J. Seaton, J. Org. Chem., 1996, 61, 6001 CrossRef CAS.
- G. Butora, T. Hudlicky, S. P. Fearnley, A. G. Gum, M. R. Stabile and K. Abboud, Tetrahedron Lett., 1996, 37, 8155 CrossRef CAS.
- D. L. J. Clive, Y. Zhou and D. P. de Lima, Chem. Commun., 1996, 1463 RSC.
- R. Clauss and R. Hunter, J. Chem. Soc., Perkin Trans. 1, 1997, 71 RSC.
- Y.-M. Tsai, H.-C. Nieh, J.-S. Pan and D.-D. Hsiao, Chem. Commun., 1996, 2469 RSC.
- M.-P. Denieul, B. Quiclet-Sire and S. Z. Zard, Tetrahedron Lett., 1996, 37, 5495 CrossRef CAS.
- M.-H. Chen and J. A. Abraham, Tetrahedron Lett., 1996, 37, 5233 CrossRef CAS.
- Y. Guindon and R. C. Denis, Tetrahedron Lett., 1998, 39, 339 CrossRef CAS.
- L. Engman and V. Gupta, J. Org. Chem., 1997, 62, 157 CrossRef CAS.
- A. Srikrishna, R. Viswajanani and C. V. Yelamaggad, Tetrahedron, 1997, 53, 10479 CrossRef CAS.
- J. Adrio and J. C. Carreterro, Tetrahedron, 1998, 54, 1601 CrossRef CAS.
- D. P. Stamos, S. S. Chen and Y. Kishi, J. Org. Chem., 1997, 62, 7552 CrossRef CAS.
- W. R. Dolbier and X. X. Rong, J. Org. Chem., 1996, 61, 5321 CrossRef.
- P. A. Baguley, G. Binmore, A. Milne and J. C. Walton, Chem. Commun., 1996, 2199 RSC.
- T. Ooi, Y. Hokke and K. Maruoka, Angew. Chem., Int. Ed. Engl., 1997, 36, 1181 CrossRef CAS.
- R. Giovannini and M. Petrini, Chem. Commun., 1997, 1829 RSC.
- U. P. Topiwala, M. C. Luszniak and D. A. Whiting, J. Chem. Soc., Perkin Trans. 1, 1998, 1185 RSC; S. P. Green and D. A. Whiting, J. Chem. Soc., Perkin Trans. 1, 1998, 193 RSC.
- A. Ogawa, R. Obayashi, H. Ine, Y. Tsuboi, N. Sonoda and T. Hirao, J. Org. Chem., 1998, 63, 881 CrossRef CAS.
- C. Anies, A. Pancrazi and J.-Y. Lallemand, Bull. Soc. Chim. Fr, 1997, 134, 183 Search PubMed.
- A. Vaupel and P. Knochel, J. Org. Chem., 1996, 61, 5743 CrossRef CAS.
- E. Riguet, I. Klement, C. K. Reddy, G. Cahiez and P. Knochel, Tetrahedron Lett., 1996, 37, 5865 CrossRef CAS.
- A. K. Ghosh, J. F. Kincaid, D. E. Walters, Y. Chen, N. C. Chaudhuri, W. J. Thompson, C. Culberson, P. M. D. Fitzgerald, H. Y. Lee, S. P. McKee, P. M. Munson, T. T. Duong, P. L. Darke, J. A. Zugay, W. A. Schleif, M. G. Axel, J. Lin and J. R. Huff, J. Med. Chem., 1996, 39, 3278 CrossRef CAS.
- P. K. Mandal, G. Maiti and S. C. Roy, J. Org. Chem., 1998, 63, 2829 CrossRef CAS; G. Chambournier, V. Krishnamurthy and V. H. Rawal, Tetrahedron Lett., 1997, 38, 6313 CrossRef CAS; P. A. Wender, T. M. Dore and M. A. deLong, Tetrahedron Lett., 1996, 37, 7687 CrossRef CAS.
- P. A. Evans and T. Manangan, Tetrahedron Lett., 1997, 38, 8165 CrossRef CAS; P. A. Evans and J. D. Roseman, Tetrahedron Lett., 1997, 38, 5249 CrossRef CAS.
- P. A. Evans, J. D. Roseman and L. T. Garber, J. Org. Chem., 1996, 61, 4880 CrossRef CAS.
- E. Lee, Y.-W. Jeong and Y. Yu, Tetrahedron Lett., 1997, 38, 7765 CrossRef CAS.
- G. A. Molander and C. R. Harris, J. Org. Chem., 1997, 62, 2944 CrossRef CAS.
- S. Hanessian, J. Pan, A. Carnell, H. Bouchard and L. Lesage, J. Org. Chem., 1997, 62, 465 CrossRef CAS.
- D. Damour, G. Doerflinger and S. Mignani, Heterocycles, 1997, 45, 911 Search PubMed.
- M. Ihara, A. Katsumata and K. Fukumoto, J. Chem. Soc., Perkin Trans. 1, 1997, 991 RSC.
- J. D. White, C. S. Nylund and N. J. Green, Tetrahedron Lett., 1997, 38, 7329 CrossRef CAS; J. D. White and H. Shin, Tetrahedron Lett., 1997, 38, 1141 CrossRef CAS; A. Katsumata, T. Iwaki, K. Fukumoto and M. Ihara, Heterocycles, 1997, 46, 605 Search PubMed; G. Pandey, K. S. Sesha Poleswara Rao and K. V. Nageshwar Rao, J. Org. Chem., 1996, 61, 6799 CrossRef CAS.
- D. L. J. Clive and J. Zhang, Chem. Commun., 1997, 549 RSC.
- O. Hamelin, J.-P. Deprés and A. E. Greene, J. Am. Chem. Soc., 1996, 118, 9992 CrossRef CAS.
- S. Fukuzawa, K. Seki, M. Tatsuzawa and K. Mutoh, J. Am. Chem. Soc., 1997, 119, 1482 CrossRef CAS.
- M. D. Bachi and E. E. Korshin, Synlett, 1998, 122 CAS.
- Y. Ushigoe, Y. Kano and M. Nojima, J. Chem. Soc., Perkin Trans. 1, 1997, 5 RSC.
- P. R. Jenkins and A. J. Wood, Tetrahedron Lett., 1997, 38, 1853 CrossRef CAS.
- A. M. Gómez, S. Mantecón, S. Valverde and J. C. López, J. Org. Chem., 1997, 62, 6612 CrossRef CAS; J. Marco-Contelles, Chem. Commun., 1996, 2629 RSC; P. M. J. Jung, J. Dauvergne, A. Burger and J.-F. Biellmann, Tetrahedron Lett., 1997, 38, 5877 CrossRef CAS; B. Noya and R. Alonso, Tetrahedron Lett., 1997, 38, 2745 CrossRef CAS; R. L. Dorta, A. Martin, J. A. Salazar and E. Suárez, Tetrahedron Lett., 1996, 37, 6021 CrossRef CAS; M. Breithor, U. Herden and H. M. R. Hoffmann, Tetrahedron, 1997, 53, 8401 CrossRef CAS; H. M. R. Hoffmann, U. Herden, M. Breithor and O. Rhode, Tetrahedron, 1997, 53, 8383 CrossRef CAS.
- G. Pandey, S. Hajra, M. K. Ghorai and K. R. Kumar, J. Org. Chem., 1997, 62, 5966 CrossRef CAS.
- M. A. Lucas and C. H. Schiesser, J. Org. Chem., 1998, 63, 3032 CrossRef CAS.
- M. J. Laws and C. H. Schiesser, Tetrahedron Lett., 1997, 38, 8429 CrossRef CAS.
- M. A. Lucas and C. H. Schiesser, J. Org. Chem., 1996, 61, 5754 CrossRef CAS; Y. Kita, M. Egi, M. Ohtsubo, T. Saiki, T. Takada and H. Tohma, Chem. Commun., 1996, 2225 RSC.
- I. Ryu, T. Okuda, K. Nagahara, N. Kambe, M. Komatsu and N. Sonoda, J. Org. Chem., 1997, 62, 7550 CrossRef CAS.
- M. L. E. N. da Mata, W. B. Motherwell and F. Ujjainwalla, Tetrahedron Lett., 1997, 38, 141 CrossRef.
- E. Bonfand, W. B. Motherwell, A. M. K. Pennell, M. K. Uddin and F. Ujjainwalla, Heterocycles, 1997, 46, 523 Search PubMed.
- C. R. A. Godfrey, P. Hegarty, W. B. Motherwell and M. K. Uddin, Tetrahedron Lett., 1998, 39, 723 CrossRef CAS; M. L. E. N. da Mata, W. B. Motherwell and F. Ujjainwalla, Tetrahedron Lett., 1997, 38, 137 CrossRef.
- L. Capella, P. C. Montevecchi and M. L. Navacchia, J. Org. Chem., 1996, 61, 6783 CrossRef CAS.
- M. Journet, A. Rouillard, D. Cai and R. D. Larsen, J. Org. Chem., 1997, 62, 8630 CrossRef CAS.
- J. Malmström, V. Gupta and L. Engman, J. Org. Chem., 1998, 63, 3318 CrossRef.
- S. Bogen and M. Malacria, J. Am. Chem. Soc., 1996, 118, 3992 CrossRef CAS.
- T. Linker, M. Maurer and F. Rebien, Tetrahedron Lett., 1996, 37, 8363 CrossRef CAS.
- S. Shuto, M. Kanazaki, S. Ichikawa, N. Minakawa and A. Matsuda, J. Org. Chem., 1998, 63, 746 CrossRef CAS; S. Shuto, M. Kanazaki, S. Ichikawa and A. Matsuda, J. Org. Chem., 1997, 62, 5676 CrossRef CAS; H. Shinokubo, K. Oshima and K. Utimoto, Bull. Chem. Soc. Jpn., 1997, 70, 2255 CAS.
- Y.-M. Tsai, K.-H. Tang and W.-T. Jiaang, Tetrahedron Lett., 1996, 37, 7767 CrossRef.
- A. P. Dobbs, K. Jones and K. T. Veal, Tetrahedron Lett., 1997, 38, 5379 CrossRef CAS; A. P. Dobbs, K. Jones and K. T. Veal, Tetrahedron, 1998, 54, 2149 CrossRef CAS.
- V. F. Patel, S. L. Andis, J. K. Enkema, D. A. Johnson, J. H. Kennedy, F. Mohamadi, R. M. Schultz, D. J. Soose and M. M. Spees, J. Org. Chem., 1997, 62, 8868 CrossRef CAS; D. L. Boger, R. M. Garbaccio and Q. Jin, J. Org. Chem., 1997, 62, 8875 CrossRef CAS.
- H. Ishibashi, H. Kawanami and M. Ikeda, J. Chem. Soc., Perkin Trans. 1, 1997, 817 RSC.
- H. Ishibashi, H. Kawanami, H. Nakagawa and M. Ikeda, J. Chem. Soc., Perkin Trans. 1, 1997, 2291 RSC.
- C.-W. Ko and T. Chou, Tetrahedron Lett., 1997, 38, 5315 CrossRef CAS.
- C.-Y. Cheng, L.-W. Hsin and J.-P. Liou, Tetrahedron, 1996, 52, 10935 CrossRef CAS; G. Butora, T. Hudlicky, S. P. Fearnley, M. R. Stabile, A. G. Gum and D. Gonzalez, Synthesis, 1998, 665 CrossRef CAS.
- B. K. Banik, V. S. Raju, M. S. Manhas and A. K. Bose, Heterocycles, 1998, 47, 639 Search PubMed.
- Y. Hayashi, H. Shinokubo and K. Oshima, Tetrahedron Lett., 1998, 39, 63 CrossRef CAS.
- I. Ryu, T. Niguma, S. Minakata, M. Komatsu, S. Hadida and D. P. Curran, Tetrahedron Lett., 1997, 38, 7883 CrossRef CAS.
- R. Inoue, J. Nakao, H. Shinokubo and K. Oshima, Bull. Chem. Soc. Jpn., 1997, 70, 2039 CAS; J. Nakao, R. Inoue, H. Shinokubo and K. Oshima, J. Org. Chem., 1997, 62, 1910 CrossRef CAS.
- D. Crich and J.-T. Hwang, J. Org. Chem., 1998, 63, 2765 CrossRef CAS.
- A. M. Rosa, A. M. Lobo, P. S. Branco, S. Prabhakar and M. Sá-da-Costa, Tetrahedron, 1997, 53, 299 CrossRef CAS; A. M. Rosa, A. M. Lobo, P. S. Branco, S. Prabhakar and A. M. D. L. Pereira, Tetrahedron, 1997, 53, 269 CrossRef CAS.
- A. M. Rosa, A. M. Lobo, P. S. Branco and S. Prabhakar, Tetrahedron, 1997, 53, 285 CrossRef CAS.
- T. C. T. Ho and K. Jones, Tetrahedron, 1997, 53, 8287 CrossRef CAS.
- O. Tsuge, T. Hatta and H. Tsuchiyama, Chem. Lett., 1998, 155 CrossRef CAS.
- A. Padwa, M. Dimitroff, A. G. Waterson and T. H. Wu, J. Org. Chem., 1998, 63, 3986 CrossRef CAS.
- F. Aldabbagh, W. R. Bowman and E. Mann, Tetrahedron Lett., 1997, 38, 7937 CrossRef CAS.
- C. J. Moody and C. L. Norton, J. Chem. Soc., Perkin Trans. 1, 1997, 2639 RSC; S.-F. Wang and C.-P. Chuang, Tetrahedron Lett., 1997, 38, 7597 CrossRef CAS.
- F. Aldabbagh and W. R. Bowman, Tetrahedron Lett., 1997, 38, 3793 CrossRef CAS.
- S. Caddick, C. L. Shering and S. N. Wadman, Tetrahedron Lett., 1997, 38, 6249 CrossRef CAS.
- T. Uetake, M. Nishikawa and M. Tada, J. Chem. Soc., Perkin Trans. 1, 1997, 3591 RSC.
- J. A. Murphy, K. A. Scott, R. S. Sinclair and N. Lewis, Tetrahedron Lett., 1997, 38, 7295 CrossRef CAS.
- R. Leardini, D. Nanni, P. Pareschi, A. Tundo and G. Zanardi, J. Org. Chem., 1997, 62, 8394 CrossRef CAS.
- M. Black, J. I. G. Cadogan, H. McNab, A. D. MacPherson, V. P. Roddam, C. Smith and H. R. Swanson, J. Chem. Soc., Perkin Trans. 1, 1997, 2483 RSC.
- R. Leardini, H. McNab, D. Nanni, S. Parsons, D. Reed and A. G. Tenan, J. Chem. Soc., Perkin Trans. 1, 1998, 1833 RSC; M. Black, J. I. G. Cadogan, R. Leardini, H. McNab, G. McDougald, D. Nanni, D. Reed and A. Zompatori, J. Chem. Soc., Perkin Trans. 1, 1998, 1825 RSC.
- G. Calestani, R. Leardini, H. McNab, D. Nanni and G. Zanardi, J. Chem. Soc., Perkin Trans. 1, 1998, 1813 RSC.
- C. M. Camaggi, R. Leardini, D. Nanni and G. Zanardi, Tetrahedron, 1998, 54, 5587 CrossRef CAS.
- A. J. Blake, G. J. Hollingworth and G. Pattenden, Synlett, 1996, 643 CrossRef CAS.
- G. Pattenden and P. Wiedenau, Tetrahedron Lett., 1997, 38, 3647 CrossRef CAS.
- A. Philippon, J. Tao, D. Tétard, M. Degueil-Castaing and B. Maillard, Synth. Commun., 1997, 27, 2651 CAS; A. L. J. Beckwith, K. Drok, B. Maillard, M. Degueil-Castaing and A. Philippon, Chem. Commun., 1997, 499 RSC.
- S. E. Gibson, N. Guillo and M. J. Tozer, Chem. Commun., 1997, 637 RSC.
- Y. Yuasa, W. Sato and S. Shibuya, Synth. Commun., 1997, 27, 573 CAS; M. Sasaki, M. Inoue, T. Noguchi, A. Takeichi and K. Tachibana, Tetrahedron Lett., 1998, 39, 2783 CrossRef CAS.
- E. Lee and C. H. Yoon, Tetrahedron Lett., 1996, 37, 5929 CrossRef CAS.
- P. Chattopadhyay, M. Mukherjee and S. Ghosh, Chem. Commun., 1997, 2139 RSC.
- M. Ikeda, K. Obata, J. Oka, H. Ishibashi and T. Sato, Heterocycles, 1997, 44, 203 Search PubMed.
- S. Hadida, M. S. Super, E. J. Beckman and D. P. Curran, J. Am. Chem. Soc., 1997, 119, 7406 CrossRef CAS.
- A. Routledge, C. Abell and S. Balasubramanian, Synlett, 1997, 61 CrossRef CAS.
- X. Du and R. W. Armstrong, Tetrahedron Lett., 1998, 39, 2281 CrossRef CAS; X. Du and R. W. Armstrong, J. Org. Chem., 1997, 62, 5678 CrossRef CAS.
- S. Ozaki, E. Matsui, J. Waku and H. Ohmori, Tetrahedron Lett., 1997, 38, 2705 CrossRef CAS.
- C.-K. Sha, K. C. Santhosh, C.-T. Tseng and C.-T. Lin, Chem. Commun., 1998, 397 RSC.
- S.-F. Wang and C.-P. Chuang, Heterocycles, 1997, 45, 347 Search PubMed.
- J. A. Murphy, F. Rasheed, S. Gastaldi, T. Ravishanker and N. Lewis, J. Chem. Soc., Perkin Trans. 1, 1997, 1549 RSC.
| This journal is © The Royal Society of Chemistry 2000 |