Thermodynamically self-assembling porphyrin-stoppered rotaxanes
Maxwell J. Gunter*a, Nick Bampos*b, Ken D. Johnstonea and Jeremy K. M. Sandersb
aDi![[italic v]](https://www.rsc.org/images/entities/char_e0f5.gif) ision of Chemistry, Uniersity of New England, Armidale, NSW 2351, Australia. E-mail: mgunter@metz.une.edu.au
ision of Chemistry, Uniersity of New England, Armidale, NSW 2351, Australia. E-mail: mgunter@metz.une.edu.au
bUniersity Chemical Laboratory, Lensfield Road, Cambridge, UK CB2 1EW. E-mail: nb10013@cam.ac.uk
First published on 1st December 2000
Abstract
A
variety of porphyrin-stoppered rotaxanes has been assembled under conditions of thermodynamic reversibility
by judicious choice of components and temperatures. An admixture of a thread unit comprising a central naphthodiimide
with terminal pyridines (2a), a ring unit dinaphtho-38-crown-10 (1) and either ZnII (3a), RuII (CO)
(3b) or RhIII I (3c) as stoppers was shown to form an equilibrating mixture of pseudorotaxanes and the porphyrin-stoppered
rotaxanes. The intact rotaxane could be crystallised from solution and chromatographed at low
temperatures; at higher temperatures only a mixture of the components separated on chromatography. 1H NMR
revealed NOE correlations between all three components for the rotaxane, and exchange peaks for related free and
complexed species. The temperature and concentration dependence of the equilibria were studied by NMR methods,
and van't Hoff plots for the Ru(CO) stoppered system enabled an estimation of ΔH° and ΔS°, −41.4 kJ mol−1,
and −95 J K−1 mol−1, respectively, and a Ka at 273 K of 790 M−1, rising to 5.39 × 104 M−1 at 223 K. For the unstoppered
pseudorotaxane systems 1–2a and 1–2b, the R ln K ![[italic v]](https://www.rsc.org/images/entities/i_char_e0f5.gif) s. 1/T plots were not linear, implying a temperature-dependent ΔH
and a non-zero ΔCp, indicative of a folding/unfolding of the extended thread unit on complexation. Nevertheless,
although the thermodynamic stability of the overall rotaxane is expected to be comparable to that of the pseudorotaxane, there is clearly an enhanced kinetic barrier for formation of the metalloporphyrin-stoppered
rotaxanes, but not for the more labile zinc analogue. While mixing of all three components of the rotaxanes at
room temperature resulted in rapid rotaxane assembly irrespective of the order of addition (thermodyamic control), it
was shown that at low temperatures it was possible to “lock out” or “lock on” the central thread unit under conditions of
kinetic control. These concepts were further extended to the assembly of more complex multi-porphyrin arrays, where the central ring unit is a naphthocrown-strapped zinc porphyrin. It is therefore possible to use the kinetics of the remote event (metal ligation/coordination)
to control
the overall kinetics
of rotaxane formation.
s. 1/T plots were not linear, implying a temperature-dependent ΔH
and a non-zero ΔCp, indicative of a folding/unfolding of the extended thread unit on complexation. Nevertheless,
although the thermodynamic stability of the overall rotaxane is expected to be comparable to that of the pseudorotaxane, there is clearly an enhanced kinetic barrier for formation of the metalloporphyrin-stoppered
rotaxanes, but not for the more labile zinc analogue. While mixing of all three components of the rotaxanes at
room temperature resulted in rapid rotaxane assembly irrespective of the order of addition (thermodyamic control), it
was shown that at low temperatures it was possible to “lock out” or “lock on” the central thread unit under conditions of
kinetic control. These concepts were further extended to the assembly of more complex multi-porphyrin arrays, where the central ring unit is a naphthocrown-strapped zinc porphyrin. It is therefore possible to use the kinetics of the remote event (metal ligation/coordination)
to control
the overall kinetics
of rotaxane formation.
Introduction
As the sophistication and complexity of supramolecular systems increase, simplicity in assembly and synthesis assumes an ever more important role; this has been matched by moves towards the production of modular components that can be assembled readily and predictably. Product selection through thermodynamic preference is a key concept in these approaches and there have been many examples where these principles have been demonstrated, such as in helicates,1 macrocyclic synthesis,2,3 thermodynamic templating for cyclic oligomer formation,4 molecular amplification in a dynamic combinatorial library,5 cyclic porphyrin dimers,6 large polyhedral capsules,7,8 and catenane formation.9–11 Porphyrin-containing systems of increasing complexity can also be assembled through self-assembling, reversible processes to produce intricate shapes and motifs, including squares,12 three-dimensional arrays and tapes,13–15 ladders,16,17 wires18 and various multi-metal assemblies;19–22 and more recently an elegant application of non-covalent assembly through a combination of hydrogen bonding and porphyrin coordination has resulted in the guest-templated selection of an intricate receptor in a thermodynamically equilibrating combinatorial mixture.23 We now illustrate how similar principles can be applied to rotaxane assembly as an efficient and rapid route to porphyrin-based multicomponent systems that have particular relevance to photophysical electron and energy-transfer systematics.24,25 We also show how the kinetics of rotaxane assembly can be influenced by remote metal complexation/decomplexation events.Pseudorotaxane
formation inherently involves non-covalent and
thus reversible assembly of the components. Conversion into
a more or less permanently interlocked rotaxane ia the stoppering
approach is usually achieved under kinetic control for
covalent attachment of the required bulky end-groups. On the
other hand, an alternative so-called slippage approach is based
on thermodynamic control with heating necessary to overcome
the forward kinetic barrier while preventing dethreading
through the reverse process with its higher activation barrier. Obviously, the success of this approach relies critically
on the size selection process that matches the dimensions
of the ring with the stoppers. This is a serious limitation for
systems in which porphyrins are to be used as potentially photo-
and electro-active stoppers. More recently, an alternative approach
to self-assembly of rotaxanes under conditions of thermodynamic
reversibility using imine chemistry has been reported;26
in the resulting rotaxane it was demonstrated that exchange
of stoppers could be effected under kinetic control without
dethreading.27 On the other hand, metal–ligand coordination
can be employed for thermodynamic control26
and this has
been a successful strategy for a variety of pseudo-rotaxane
and rotaxane-like structures, either as a method for
attaching
stoppers in cyclodextrin-based28–30 and other systems,31,32
or for assembling the ring unit around a pre-formed thread
in a clipping approach.33 Sauvage and co-workers have extensively
used templating metal ions around a central coordination
site to assemble porphyrin-stoppered rotaxanes and catenanes.34–37
Nevertheless, the stoppering of a pre-formed pseudorotaxane
ia porphyrin axial coordination chemistry had
not been described until very recently, when Branda and co-workers38
reported such a system at the same time that we had
independently been investigating similar strategies.
However, in all of these processes, the distinction between rigorously defined terms rotaxane and pseudorotaxane becomes blurred, and to some extent such a definition requires inclusion of a time-frame; these factors have been recognised previously,39 where for example a pseudorotaxane in solution can be considered a rotaxane in its corresponding solid state structure. It is also interesting that for the ionic rotaxane structures reported by Branda and co-workers38 there was an added complication of a solvent dependence of the equilibrium constant for the pseudorotaxane, but no corresponding values were reported for the porphyrin-stoppered system. Hence no comparison of the thermodynamic parameters for the two systems was presented, and the solution state dynamics was not discussed. We intended to investigate more fully the solution dynamics of the self-assembly process of rotaxanes using axial coordination chemistry of a variety of metalloporphyrins as stoppers, and the results are reported here.
Results and discussion
For a modular thermodynamic approach to self-assembled rotaxane structures the equilibria in Scheme 1 need to be considered, where both threading and stoppering are reversible processes. We have chosen components with dimensions such that it can reasonably be assumed that K2′ and K2 (coordination at the termini of the thread unit) would not be significantly different; this implies the absence of any cooperativity or repulsive interactions between coordination of the porphyrin stoppers within the pseudorotaxane structure compared to the uncomplexed thread.40 In this case it can easily be shown by manipulation of the equilibrium expressions that the overall formation constant K3 = K1. Nevertheless, the kinetic barriers for each process will be very different, as it is clear that direct complexation/decomplexation of the pre-stoppered thread cannot take place, and can only occuria the stepwise
process of coordination/decoordination of the thread
and the metalloporphyrin; this is a classic case of the distinction
between thermodynamic stability s. kinetic lability, and
is illustrated schematically in Fig. 1. | ||
| Scheme 1 | ||
 | ||
| Fig. 1 Schematic energy profiles for the processes depicted in Scheme 1, illustrating the essential differences between the Zn, Ru(CO), and RhI porphyrin stopper units in the assembly of the rotaxanes 6. | ||
For our purposes we chose a thread 2a with a naphthodiimide as a central recognition component and pyridines as terminal ligands,41 while the ring 1 was the naphthocrown ether dinaphtho-38-crown-10 (DN38-C-10). 2a was synthesized from the corresponding substituted naphthodiimide and nicotinic acid chloride.41 It has been well established that neutral diimides of the type 2 bind effectively to DN38-C-10 through a combination of electrostatic, hydrogen-bonding and donor–acceptor interactions resulting from π–π stacking, and these components have been employed for a variety of interlocked systems.9,10,41,42 For the detachable stoppers 3 the choice was between zinc 3a, RuII(CO) 3b and RhIIII 3c complexes of the readily soluble and sterically demanding meso-5,10,15,20-tetrakis(3,5-di-tert-butylphenyl)porphyrin. Thus the target rotaxanes were 6a–6c.

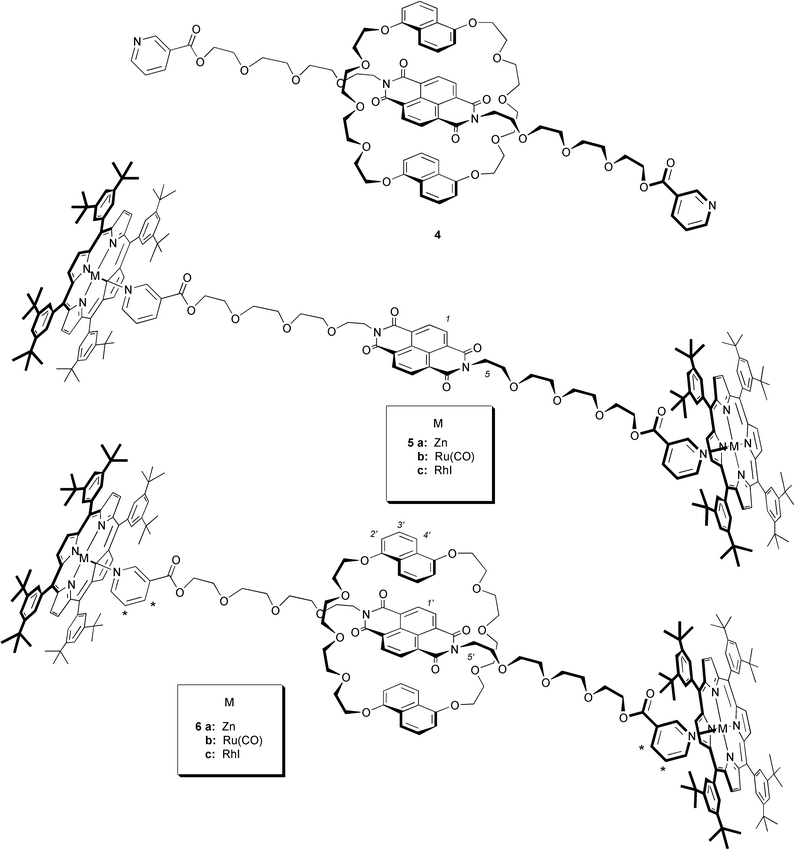
Zinc porphyrins form five-coordinate labile complexes with nitrogen donor ligands, while the six-coordinate Ru(CO) and RhI nitrogen donor complexes are comparatively much more stable and inert.43 For typical pyridine zinc meso-tetraarylporphyrins at room temperature, ligand exchange is fast on the NMR chemical shift timescale, with on-rates of more than 108 M s−1 and off-rates around 105 s−1, giving an overall Ka (K2′ in Scheme 1) of 103–104 M−1. On the other hand ruthenium(II) carbonyl porphyrins exhibit a rapid on-rate and slow off-rate (ca. 0.1 s−1) with Ka values of 107 to 108 M−1 for typical pyridine ligands, and exchange is slow on the NMR timescale. Rhodium(III) iodo complexes are more stable still, with Ka estimated at >109 M−1.43 By comparison, the stability constants (K1 in Scheme 1) for naphthodiimide binding to dinaphtho-38-crown-10 are of the order of 103 M−1,42 and this exchange is rapid on the NMR timescale at room temperature. Thus, in the first instance, variation of the metalloporphyrin stoppers while maintaining constant DN38-C-10 and naphthodiimide thread components 1 and 2a allowed us to establish a useful basis set for timescale and temperature dependence of the overall rotaxane self-assembly process, K3 (Scheme 1).
Simply mixing the components in the required stoichiometry for rotaxane formation at room temperature in deuteriochloroform at millimolar concentrations produced a new set of peaks in the NMR spectrum, distinct from those of the individual components (Fig. 2). The same result was obtained irrespective of the order of the addition, and the spectra did not change over time when the solutions were maintained at ambient temperature. However, variation in the temperature or the overall concentration changed the relative ratios of the peaks, which is consistent with a dynamic and rapid equilibration. The results of two-dimensional NMR experiments (COSY, NOESY and C–H correlation) were consistent with the new set of upfield shifted peaks arising from the rotaxane structure 6, and both exchange-related and coupled peaks indicated an equilibrium with the individual components. In the case of the Zn–porphyrin stoppered systems all components (thread, stoppers, ring, unstoppered pseudorotaxane and intact rotaxane) were observed in fast exchange on the NMR chemical shift timescale at room temperature, whereas the Ru(CO) and RhI analogues showed only stoppered thread 5b and 5c, ring 1, and rotaxane 6b and 6c in equilibrium (reflecting the large stability constant of the pyridine ligands for these metallo-derivatives; for a 1:2 bidentate ligand:metalloporphyrin ratio at these concentrations, more than 99% of the ligand is coordinated).
 | ||
| Fig. 2 Partial 1H NMR (400 MHz) spectrum of an admixture of compounds 1, 2a and 3b (1:1:2 molar ratio) in CDCl3 at 0 °C. Numbered peaks are those of the uncomplexed 1 and coordinated thread component 5b, and the dashed numbers are their corresponding positions in the rotaxane 6b. Larger unnumbered peaks are for porphyrin β-pyrrole and meso-aryl (S is solvent). The peaks between δ 3 and 4 are due to crown ether methylenes. Peaks 1–5 are due to naphthodiimide, DN38-C-10 and crown methylenes, respectively, as indicated in the structures. Peaks marked with asterisks (*) are assigned to H4 and H5 of the coordinated pyridines (H6 and H2 appear at δ 2.21 and 1.76, respectively). | ||
Nevertheless, in all cases the intact rotaxane could be crystallised from solution by slow infusion of methanol into chloroform solutions of the admixed components. Although at this stage none of the crystals has yielded to X-ray structural analysis, redissolution of the isolated needle-like crystals reproduced the original NMR spectra of the equilibrating mixture in an identical stoichiometric ratio; neither the thread nor the ring crystallises independently under these conditions. It is also interesting that Branda and co-workers were able to isolate single crystals of X-ray quality of a related rotaxane, albeit with quite different thread, ring and stopper components.38
Variable temperature 1H NMR experiments were particularly enlightening. In all cases as the temperature was lowered the peaks due to the rotaxane grew at the expense of those of the individual components. Equilibrium was established fairly rapidly down to about 0 °C for the RhI derivatives, and about −20 °C for the Ru(CO) derivatives, but was noticeably slower at lower temperatures; it remained rapid for the zinc derivatives down to −65 °C. For the slowly exchanging systems it was possible to determine the Ka as a function of temperature, and thus in principle thermodynamic parameters from van't Hoff plots. For the Ru(CO) stoppered system 6b the linear relationship between R ln K and 1/T over the available temperature range enabled an estimation of ΔH° and ΔS°, −41.4 kJ mol−1, and −95 J K−1 mol−1, respectively, and a Ka at 273 K of 790 M−1, rising to 5.39 × 104 M−1 at 223 K. Comparable values were to be expected for the simple unstoppered complex of 1· 2a. However, although the Ka at 273 K was 448 M−1, a van't Hoff plot for this system was not linear (Fig. 3) and a direct comparison of the ΔG° and ΔS° values with those of 6b was not possible. Such a non-linear dependence is indicative of a temperature-dependent ΔH, and hence a non-zero heat capacity term ΔCp . A negative ΔCp is typically indicative of temperature dependent solvation of polar functional groups in a host and/or guest,44,45 whereas a positive value generally accompanies phenomena such as protein folding or dissolution of organic molecules in water.44,46,47 Given the conditions of these experiments (water-free CDCl3 solvent), it is reasonable to assume that the ΔCp term in this instance is positive, and reflects perhaps an inter- or intra-molecular interaction (such as π–π stacking) of the terminal pyridine groups on the thread with the dinaphtho crown and its included central diimide moiety. Clearly, such an interaction is prohibited in 6 when the pyridines are axially coordinated to the ruthenium porphyrin. We reasoned that a test for these types of interactions would be to replace the nicotinoyl moieties with non-aromatic acetyls, in compound 2b. However, contrary to our expectations, the acetyl derivative 2b showed virtually identical behaviour to that of the nicotinyl 2a (Fig. 3c). Thus we are led to conclude that the non-zero ΔCp term arises from a more subtle, but undefined, effect as a result of the rather flexible tetra(ethyleneoxy) arms, which is removed when these are restricted by terminal coordination to the ruthenium porphyrin stoppers.48
 | ||
| Fig. 3 van't Hoff plots for (a) the Ru(P) stoppered rotaxane/pseudorotaxane system 6, (b) the pseudorotaxane system resulting from an equimolar mixture of compounds 1 and 2a, and (c) the pseudorotaxane from 1 and 2b. The symbols are the measured data, and the solid line in (a) represents the fit for ΔH° = −41.4 kJ mol−1 and ΔS° = −95 J K−1 mol−1; in (b) and (c) the solid lines are a guide to the eye. | ||
As the unstoppered pseudorotaxane 1·2a exhibits fast exchange at room temperature, it was possible to extract the free enthalpy of activation for the complexation from the coalescence temperature TC, 290 K, so that ΔG‡ = 58 ± 1 kJ mol−1. On introduction of the Ru–porphyrin stoppers in 6 the system now exhibits slow exchange at room temperature, and no coalescence could be observed up to the temperature limit of the solvent (320 K). Thus although it was not possible to determine a corresponding ΔG‡ for the exchange process here, clearly it must be significantly higher than about 65 kJ mol−1, consistent with the kinetic barrier introduced by the stoppering. On the other hand, for the zinc porphyrin stoppered system which remains fast at room temperature, ΔG‡ is constant (59 ± 1 kJ mol−1), which is to be expected for such a system where the complexation and coordination rates (and the Ka) are of the same order of magnitude.
As further evidence for discrete rotaxane formation in solution, the 1H NMR spectra of compound 6b exhibited significant NOE connectivities, among others, between the diimide aromatic and N–CH2 protons and those of the crown aromatic rings and adjacent methylenes. In addition for the equilibrating mixture at 30 °C, NOE cross-peaks could only be detected within the resonances for the rotaxane, and not between the uncomplexed components; instead, these showed exchange peaks with their rotaxane counterparts. No NOE cross-peaks were detected between any of the porphyrin stopper resonances and those of the ring, indicating that the stopper units are remote from the central ring, and that the molecule most likely adopts an extended conformation in solution.
The temperature-dependent NMR behaviour suggested that it might be possible to perform experiments at low temperatures either to form the rotaxane more or less irreversibly and in high yield by prior threading and then stoppering (“locking on”), or alternatively to “lock out” rotaxane formation by stoppering before threading. This relies on the principle that K1 for complexation of the diimide within the dinaphthocrown is significantly smaller than that for coordination of the ligands with the metal ion (K2 or K2′ ), or alternatively that there is a significant difference in the limiting off-rates for the two processes. This is clearly the case for the Ru(CO) and RhI derivatives, but not necessarily so for that of Zn.
In the first instance it could be shown that the Ru(CO) and RhI rotaxanes can be chromatographed intact at low temperatures. By applying a stoichiometric mixture of the com ponents to a 2-D TLC plate, and thence developing the plate in a suitable solvent (10% MeOH in CH2Cl2) at −30 °C (in a freezer compartment), a major spot for the rotaxane could clearly be identified, in addition to smaller spots for each of the components. Re-development in an orthogonal direction, but at ambient temperature, showed spots only for the individual components, with no rotaxane. This is shown in Fig. 4.49 On the other hand, the system utilising the zinc porphyrin stoppers could not be chromatographed intact, and only the individual components separated even at −40 °C.
 | ||
| Fig. 4 The two dimensional TLC experiment described in the text. A stoichiometric mixture (1:1:2 mol ratio) of components 1, 2a and 3b was applied to the silica-coated plate at the start. The plate was cooled to −30 °C and developed at this temperature in a pre-cooled CH2Cl2–MeOH (10:1) solvent mixture. Subsequently, the plate was developed in an orthogonal direction in the same solvent mixture at +25 °C. The spots for 1 and 2a were visualised under UV light, and are traced, while the rotaxane and ruthenium porphyrin spots were visible to the naked eye. | ||
This temperature dependence was probed spectroscopically. For an increased yield of rotaxane, an equimolar mixture of the thread 2a and the ring 1 was cooled to −40 °C and maintained at this temperature while a pre-cooled solution of the appropriate metalloporphyrin was added (“locked-on” experiment). After thorough mixing, the sample was rapidly transferred without warming to an NMR spectrometer kept at −40 °C. The spectrum was then monitored as the temperature was stepped up to +30 °C, and as it was re-cooled to −40 °C. The results are illustrated in Fig. 5, where it is shown that rotaxane construction by prior threading, and subsequently stoppering at low temperatures, resulted in a significantly increased yield, more so than the fully equilibrated mixture (shown by the warmed, then re-cooled sample). Thus, by enhancing complexation of 1 and 2a by use of low temperatures, and then “locking on” the thread by even stronger coordination of the stoppers at a temperature where the off-rate of the ligand-to-metal bonding was sufficiently slow, a high yield of rotaxane 6 could be achieved. Note, however, that in the zinc case 6a, equilibration is still rapid at −40 °C, and the same equilibrium distribution is obtained whether the reaction is performed at −40 or at +30 °C and then cooled.
 | ||
| Fig. 5 Normalised
relative ratios of rotaxane s. uncomplexed components 1 and 5 for the two experiments described in the text. The hatched bars represent the proportion of rotaxane formed at −40 °C: in the case of the “locked on” experiment by mixing 1 and 2a, followed by 3b at −40 °C, and in the “locked out” case by mixing 2 and 3b, followed by 1 at −40 °C. The solid bars are the relative ratios of the same mixtures equilibrated at +30 °C followed by re-cooling to −40 °C. | ||
As a corollary to this procedure, a “locking out” experiment was performed, and the results are also shown in Fig. 5. In this case the ligand thread and the metalloporphyrin were mixed and pre-cooled, thus ensuring a “fixed” stoppered thread 5. Now addition of the ring 1 at −40 °C resulted in only minimal production of rotaxanes 6b and 6c. Again, warming to +30 °C and re-cooling resulted in the same equilibrated mixture as before. Equilibration began at a reasonable rate at about −20 °C for the Ru(CO) derivative, whereas for the RhI analogue this did not happen until about 0 °C, consistent with the previous observations. As previously, only the Ru(CO) and RhI stoppers showed this behaviour; the zinc stopper is still equilibrating too fast at −40 °C for effective “locking out” in the case of 6a.
As
a further indication of the utility of this procedure for
the ready assembly of complex systems under reversible conditions
we were able to investigate the properties of the more
complex assemblies 8, containing zinc, free-base, ruthenium and
rhodium porphyrins with a potential communication pathway
between them ia the naphthodiimide and the thread. Therefore, in place of the DN38-C-10 ring, we substituted
the naphthoquinol-strapped zinc or free-base porphyrin 7,50 and
in a similar manner by simply mixing the components we could easily establish the existence of the various rotaxanes
8 in solution. In all cases at low temperatures at millimolar concentrations it was apparent that the rotaxane was the
major component. In principle, it should now be possible
to investigate the photophysical behaviour of such systems
at low temperatures. Furthermore, it is a simple matter
of varying the components by judicious choice of metal ion for
both the central and stopper components, for a ready entry
into a regular series of differently metallated species for a systematic
study of their appropriate properties.

Conclusion
By appropriate choice of components, we have shown that it is feasible to fine tune the conditions necessary for porphyrin-stoppered rotaxane self-assembly by simple mixing of the constituent parts. It is clear that this choice can be extended to a range of threads, rings and stoppers where the appropriate combinations can be guided by the relative values of the kinetic and stability constants, and we have applied these procedures for the synthesis of related rotaxanes where the ring unit is a crown-strapped metalloporphyrin. Furthermore, the principles established in the reversible and thermodynamically controlled assembly of these rotaxanes should facilitate the construction of more complex structures, and indeed may form the basis for combinatorial libraries of supramolecular interlocked structures.Experimental
1H NMR spectra were recorded in CDCl3 solutions on Bruker AM or DRX-400 and DRX-500 and 800 MHz spectrometers using standard pulse sequences, and the residual solvent peak as reference. All spectra were assigned using a combination of COSY, NOESY, ROESY (rotating frame Overhauser enhancement spectroscopy) and C–H correlation techniques. Concentrations were typically 10–15 mM. Measurements at various temperatures were performed after at least 5 minutes equilibration (longer for lower temperatures). As a check that equilibria had been established, representative samples were held for up to 2 hours at the desired temperature, and the spectra periodically monitored. It was confirmed that no further change had taken place after this time. Additionally, sample spectra were checked for consistency after approaching the same temperature from either higher or lower values.For spectra showing slow exchange, K values were calculated from integrated values of the separate peaks and averaged over several values. Uncertainty in K values is estimated at ±10%. Rate constants for the exchange process at the coalescence temperature were calculated using standard methods.51
Low temperature experiments were performed by mixing pre-cooled solutions and the resulting mixture was also maintained at the desired temperature in an acetone–solid CO2 bath. For NMR experiments, pre-cooled solutions in NMR tubes were rapidly transferred without warming to the NMR probe previously cooled to the desired temperature.
Low temperature TLC experiments were conducted by applying the stoichiometric mixture of the components at room temperature, then cooling the plate to −35 °C in a freezer, and finally developing the plate in a pre-cooled solvent mixture (10% methanol in chloroform) in a tank in the same freezer. Colourless components were visualised under UV light.
We were unable to obtain suitable mass spectra of the intact rotaxanes 6, using FAB, electrospray or matrix-assisted laser desorption–ionisation (MALDI) techniques; in all instances only the components were identified, and in most cases even the coordinated thread units 5 were not identified, but only the unligated metalloporphryins.
Syntheses
Dinaphtho-38-crown-10 1 was synthesized as previously described,52 as were the zinc, Ru(CO) and RhI derivatives of meso-5,10,15,20-tetrakis(3,5-di-tert-butylphenyl)porphyrin.43 Compounds 2a and 2b were available from treatment of the corresponding hydroxy compound41 with either nicotinoyl chloride (for 2a) or acetyl chloride (for 2b) in excess in dry tetrahydrofuran containing at least a 10-fold excess of triethylamine. Aqueous work-up afforded both as viscous oils, which were purified further by column chromatography (SiO2, eluting with 5% MeOH in CH2Cl2). Both samples were shown to be pure by TLC in at least two different solvent systems.Spectra
Acknowledgements
This research was supported by the Australian Research Council, and a Royal Society of Chemistry Grant for International Authors (M. J. G. as visiting researcher to JKMS at the University Chemical Laboratory, Cambridge). We thank James Redman for advice and assistance with the synthesis of the rhodium porphyrins. We also thank a referee for suggestions for Fig. 1.References and notes
- B. Hasenknopf, J.-M. Lehn, N. Boumediene, A. Dupont-Gervais, A. Van Dorsselaer, B. Kneisel and D. Fenske, J. Am. Chem. Soc., 1997, 119, 10956 CrossRef CAS.
- S. J. Rowan, D. G. Hamilton, P. A. Brady and J. K. M. Sanders, J. Am. Chem. Soc., 1997, 119, 2578 CrossRef CAS.
- H. Hioki and W. C. Still, J. Org. Chem., 1998, 63, 904 CrossRef CAS.
- P. A. Brady and J. K. M. Sanders, J. Chem. Soc., Perkin Trans. 1, 1997, 3237 RSC.
- R. L. E. Furlan, G. R. L. Cousins and J. K. M. Sanders, Chem. Commun., 2000, 1761 RSC.
- G. Kaiser and J. K. M. Sanders, Chem. Commun., 2000, 1763 RSC.
- N. Takeda, K. Umemoto, K. Yamaguchi and M. Fujita, Nature (London), 1999, 398, 794 CrossRef CAS.
- B. Olenyuk, J. A. Whiteford, A. Fechtenkötter and P. J. Stang, Nature, 1999, 398, 796 CrossRef CAS.
- A. C. Try, M. M. Harding, D. G. Hamilton and J. K. M. Sanders, Chem. Commun., 1998, 723 RSC.
- D. G. Hamilton, N. Feeder, S. J. Teat and J. K. M. Sanders, New J. Chem., 1998, 22, 1019 RSC.
- T. J. Kidd, D. A. Leigh and A. J. Wilson, J. Am. Chem. Soc., 1999, 121, 1599 CrossRef CAS.
- R. V. Sloane and J. T. Hupp, Inorg. Chem., 1997, 36, 5422 CrossRef CAS.
- C. M. Drain and J.-M. Lehn, J. Chem. Soc., Chem. Commun., 1995, 2313 RSC.
- C. M. Drain, K. C. Russell and J.-M. Lehn, Chem. Commun., 1996, 337 RSC.
- C. M. Drain, F. Nifiatis, A. Vasenko and J. D. Batteas, Angew. Chem., Int. Ed., 1998, 37, 2344 CrossRef CAS.
- P. N. Taylor and H. L. Anderson, J. Am. Chem. Soc., 1999, 121, 11538 CrossRef CAS.
- H. L. Anderson, Inorg. Chem., 1994, 33, 972 CrossRef CAS.
- H. L. Anderson, Chem. Commun., 1999, 23, 2323 RSC.
- C. C. Mak, D. Pomeranc, M. Montalti, L. Prodi and J. K. M. Sanders, Chem. Commun., 1999, 21, 1083 RSC.
- H. J. Kim, J. E. Redman, M. Nakash, N. Feeder, S. J. Teat and J. K. M. Sanders, Inorg. Chem., 1999, 38, 5178 CrossRef CAS.
- H. J. Kim, N. Bampos and J. K. M. Sanders, J. Am. Chem. Soc., 1999, 121, 8120 CrossRef CAS.
- S. L. Darling, P. K. Y. Goh, N. Bampos, N. Feeder, M. Montalti, L. Prodi, B. F. G. Johnson and J. K. M. Sanders, Chem. Commun., 1998, 21, 2031 RSC.
- M. C. Calama, P. Timmerman and D. N. Reinhoudt, Angew. Chem., Int. Ed., 2000, 39, 755 CrossRef CAS.
- H. Ogoshi, T. Mizutani, T. Hayashi and Y. Kuroda, in The Porphyrin Handbook, eds. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, 2000, vol. 6, p. 279. Search PubMed.
- J. Chou, M. E. Kosal, H. S. Nalwa, N. A. Rakow and K. S. Suslick, in The Porphyrin Handbook, eds. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, 2000, vol. 6, p. 43. Search PubMed.
- S. J. Rowan and J. F. Stoddart, Org. Lett., 1999, 1, 1913 CrossRef CAS.
- S. J. Rowan and J. F. Stoddart, J. Am. Chem. Soc., 2000, 122, 164 CrossRef CAS.
- K. Yamanari and Y. Shimura, Bull. Chem. Soc. Jpn., 1984, 57, 1596 CAS.
- H. Ogino, J. Am. Chem. Soc., 1981, 103, 1303 CrossRef CAS.
- R. S. Wylie and D. H. Macartney, J. Am. Chem. Soc., 1992, 114, 3136 CrossRef CAS.
- D. Whang and K. Kim, J. Am. Chem. Soc., 1997, 119, 451 CrossRef CAS.
- R. B. Hannak, G. Färber, R. Konrat and B. Kräutler, J. Am. Chem. Soc., 1997, 119, 2313 CrossRef CAS.
- K.-S. Jeong, J. S. Choi, S.-Y. Chang and H.-Y. Chang, Angew. Chem., Int. Ed., 2000, 39, 1692 CrossRef CAS.
- N. Solladie, J. C. Chambron and J. P. Sauvage, J. Am. Chem. Soc., 1999, 121, 3684 CrossRef CAS.
- M. J. Blanco, M. C. Jimenez, J. C. Chambron, V. Heitz, M. Linke and J. P. Sauvage, Chem. Soc. Re., 1999, 28, 293 Search PubMed.
- N. Armaroli, V. Balzani, J. P. Collin, P. Gavina, J. P. Sauvage and B. Ventura, J. Am. Chem. Soc., 1999, 121, 4397 CrossRef CAS.
- P. L. Vidal, B. Divisia-Blohorn, G. Bidan, J. M. Kern, J. P. Sauvage and J. L. Hazemann, Inorg. Chem., 1999, 38, 4203 CrossRef CAS.
- K. Chichak, M. C. Walsh and N. R. Branda, Chem. Commun., 2000, 847 RSC.
- P. R. Ashton, I. Baxter, M. C. T. Fyfe, F. M. Raymo, N. Spencer, J. F. Stoddart, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1998, 120, 2297 CrossRef CAS.
- K2 and K2′ are in effect an average for two stepwise metalloporphyrin coordinations. It is reasonably assumed here that these are independent, and that there is no cooperativity. Nevertheless, confirmatory experiments with a hetero-functional thread unit were not carried out in this investigation..
- J. G. Hansen, N. Feeder, D. G. Hamilton, M. J. Gunter, J. Becher and J. K. M. Sanders, Org. Lett., 2000, 2, 449 CrossRef CAS.
- D. G. Hamilton, N. Feeder, L. Prodi, S. J. Teat, W. Clegg and J. K. M. Sanders, J. Am. Chem. Soc., 1998, 120, 1096 CrossRef CAS.
- J. K. M. Sanders, N. Bampos, Z. Clyde-Watson, S. L. Darling, J. C. Hawley, H.-J. Kim, C. C. Mak and S. J. Webb, in The Porphyrin Handbook, eds. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, New York, 2000, vol. 3, ch. 15, p. 1. Search PubMed.
- T. Hayashi, T. Miyahara, N. Koide, Y. Kato, H. Masuda and H. Ogoshi, J. Am. Chem. Soc., 1997, 119, 7281 CrossRef CAS.
- R. P. Bonar-Law and J. K. M. Sanders, J. Am. Chem. Soc., 1995, 117, 259 CrossRef CAS.
- D. A. Staufer, R. E. Barrans and D. A. Dougherty, J. Org. Chem., 1990, 52, 2762 CrossRef.
- M. J. Blandamer, J. Burges, R. E. Robertson and J. M. W. Scott, Chem. Re., 1982, 82, 259 Search PubMed.
- We were unable to detect any NOE or exchange phenomena between the terminal pyridine units and the crown, either in the pseudorotaxanes or the rotaxanes..
- This experiment was inspired by a similar technique described in T. J. Blake, J. R. Kalman and D. H. Williams, Tetrahedron Lett., 1977, 2621. Search PubMed.
- M. J. Gunter, D. C. R. Hockless, M. R. Johnston, B. W. Skelton and A. H. White, J. Am. Chem. Soc., 1994, 116, 4810 CrossRef CAS.
- H.-J. Schneider and A. Yatsimirsky, Principles and Methods in Supramolecular Chemistry, John Wiley and Sons, New York, 2000, ch. D, p. 137. Search PubMed.
- D. G. Hamilton, J. E. Davies, L. Prodi and J. K. M. Sanders, Chem. Eur. J., 1998, 4, 608 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2001 |