Making very short O–H···Ph hydrogen bonds: the example of tetraphenylborate salts
Thomas Steiner*a, Antoine M. M. Schreursb, Martin Lutzb and Jan Kroonb
aInstitut für Chemie—Kristallographie, Freie Uni![[italic v]](https://www.rsc.org/images/entities/char_e0f5.gif) ersität Berlin, Takustr. 6, D-14195, Berlin, Germany. E-mail: steiner@chemie.fu-berlin.de
ersität Berlin, Takustr. 6, D-14195, Berlin, Germany. E-mail: steiner@chemie.fu-berlin.de
bBijoet Center for Biomolecular Research, Department of Crystal and Structural Chemistry, Utrecht Uniersity, Padualaan 8, 3584 CH, Utrecht, The Netherlands
First published on 27th November 2000
Abstract
In crystal structures of tetraphenylborate salts with cationic X–OH donors (choline, triethanolammonium dihydrate, 6-ammonio-n-hexanoic acid monohydrate), the shortest intermolecular O–H···Ph hydrogen bonds known as yet have been found, both in terms of the H···M as well as the O···M distances (2.17 and 3.07 Å, respectively, for normalized O–H distance; M = midpoint of the aromatic ring). In these interactions the donor H and O atoms reside roughly over the centroids of the acceptor groups. A supporting database study of very short O–H···Ph hydrogen bonds has also been made (using the Cambridge Structural Database).
Hydrogen bonds with electrons in π bonds acting as the acceptor are attracting much current interest, both in the fields of structural chemistry and structural biology.1 Most important are those with the face of phenyl rings, X–H···Ph (‘aromatic hydrogen bonds’), which occur in many chemical and biological systems. Such bonds in the solid state have been found with a variety of donors (O–H and N–H,1 Cl–H,2 S–H,3 acidic C–H4), and their geometrical characteristics in crystals have been described with statistical methods.5 The role of X–H···Ph hydrogen bonds in heterogeneous arrays of intermolecular interactions has been analysed for the archetypical example of the aminophenols.6 Functional importance in biological recognition and as a fine-tuning mechanism in enzymatic processes is documented for a number of examples,7 and occurrence in biomolecular hydration could also be shown.8
A model system that is particularly rich in X–H···Ph hydrogen bonds is the ammonium tetraphenylborate salts, RnN+H4-n·Ph4B−.9–12 The Ph4B− anion carries four phenyl rings with a total of eight aromatic faces as potential hydrogen bond acceptors. The negative charge is not localized at the central B atom, but is diluted over the whole ion; computations at the 6-31G* level obtained fractional charges of about 0.23 e on B and 0.19 e on each phenyl ring.9 This makes the Ph groups in Ph4B− stronger hydrogen bond acceptors than phenyl rings in uncharged compounds. Therefore, it is not surprising that the shortest aromatic hydrogen bonds known occur in tetraphenylborates, with the extreme example in NH4+·Ph4B− examined by neutron diffraction (N···M distance at 15 K = 3.023 Å, where M is the aromatic centroid).12
There is a large bulk of experimental material on ‘normal’ aromatic hydrogen bonds with X···M distances in the range 3.2–3.6 Å, whereas the short distance end of the interaction is only poorly investigated. This lack of data is particularly pronounced for O–H donors. This is unfortunate because, for hydrogen bonds in general, the shortest variants of a kind X–H···A are typically of particular chemical interest, and carry information that is important for the entire family. Exploring the short distance end of N/O–H···Ph hydrogen bonding is of relevance for understanding the properties of aromatic hydrogen bonding on the whole. Since Ph4B− is an exceptionally good acceptor for N+–H donors, and also has been used successfully for the preparation of C–H···Ph hydrogen bonds,11,13 it is a promising candidate to study the short distance region also of O–H···Ph interactions. We have therefore prepared and studied a series of Ph4B− salts with cations carrying hydroxy groups, with the goal to obtain short O–H···Ph hydrogen bonds. The aim has been not only to obtain new structural data, but also to explore more generally the possibilities to make O–H···Ph hydrogen bonds with desired properties (here: short distance) on purpose.
Experimental
Samples
Tetraphenylborate salts were prepared following literature procedures.9 Aqueous solutions of Na+·Ph4B− (Lancester) and chloride salts of the target cations were mixed in equimolar ratios. The precipitates formed were filtered off, washed with water, dried, and recrystallized by slow evaporation of solutions in acetone. Crystals of sufficient quality for diffraction experiments could be obtained for the cations choline (chloride salt from Sigma), triethanolammonium (chloride salt from Merck) and 6-ammonio-n-hexanoic acid (chloride salt prepared by slow evaporation of a HCl solution of 6-amino-n-hexanoic acid from Sigma). The general observation was made that this method is more capricious with cations carrying hydroxy or carboxylic acid groups than with pure ammonium ions, for which it is very straightforward.9 For a number of interesting cations, no tetraphenylborate salts could be obtained this way (cations of 3-hydroxypyridine, glycine, histidine, methylhistidine, lysine, cysteine, arginine, glycylglycine, carnosine, glucosamine). At least for some of the examples, this could be due to the sensitivity of the tetraphenylborate ion which may suffer attack on the B–C bonds. For some other examples, crystalline tetraphenylborate salts could be obtained, but crystal structures were so heavily disordered that hydrogen bonding could not properly be assigned (Ph4B salts of N-ethyl-3-hydroxy pyridine and 2-ammonio-2-methyl-n-propane-1,3-diol).Crystallography
X-Ray diffraction data were collected at cryogenic temperature (ca. 125 K) on a Nonius Kappa–CCD diffractometer with graphite-monochromated Mo-Kα radiation (λ = 0.710 73 Å) to θmax = 27.5°. For compound 1 only twinned crystals were found in the sample, and finally diffraction data on such a crystal were collected for both lattice orientations (twin operation: twofold rotation around [3 0 1]). The crystal structures were solved and refined with standard methods14 (no absorption correction, anisotropic refinement on F2 of all reflections, hydrogen atoms bonded to C treated in the riding model with the default bond length at the temperature of measurement, hydrogen atoms bonded to N and O located in Fourier difference calculations and refined isotropically). The twin refinement for 1 was performed with SHELXL 97,14b using data in HKLF5 format only to θmax = 25.0°, and refining batch scale factors. The data are not merged in this kind of refinement, so that the number of independent reflections equals the number of measured reflections. Relevant crystal and refinement data are given in Table 1.| 1 | 2 | 3 | |
|---|---|---|---|
| a In the twin refinement the data were not merged. | |||
| Anion | Choline | Triethanolammonium | 6-Ammonio-n-hexanoic acid |
| Formula | C5H14NO+C24H20B− | C6H16NO3+C24H20B−·2H2O | C6H14NO2+C24H20B−·H2O |
| M | 423.38 | 505.44 | 469.41 |
| Crystal system | Monoclinic | Triclinic | Monoclinic |
| Space group | P21/c (no. 14) | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2) (no. 2) | C2/c (no. 15) |
| Z | 4 | 2 | 8 |
| a/Å | 9.743(2) | 11.2759(6) | 33.832(16) |
| b/Å | 13.474(7) | 11.6163(4) | 10.11(2) |
| c/Å | 18.902(11) | 13.0526(6) | 17.45(4) |
| α/° | 69.066(3) | ||
| β/° | 107.08(3) | 70.612(2) | 117.750(2) |
| γ/° | 61.747(3) | ||
| V/Å3 | 2372(2) | 1378.8(1) | 5283(16) |
| Measured reflections | 44261 | 13145 | 27080 |
| Independent reflections | 44261a | 6323 | 5947 |
| R[I>2σ(I)] | 0.0871 | 0.0511 | 0.0553 |
| wR(F2) | 0.1811 | 0.1121 | 0.1250 |
CCDC reference number 440/224. See http://www.rsc.org/suppdata/nj/b0/b004932h/ for crystallographic files in .cif format.
Results and discussion
Choline tetraphenylborate, 1
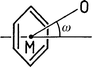
Choline, Me3N+C2H4OH, is an obviously interesting cation in the present context. Since the ammonium group is quarternary, the hydroxy group does not have to compete with any hydrogen bond donors of similar or greater strength. In the case that salt formation with Ph4B− occurs without cocrystallization of water molecules, dominant O–H···Ph hydrogen bonding can be expected. In the crystal structure of choline tetraphenylborate, 1, this expectation is indeed fulfilled. The hydroxyl group points almost directly into the face of a phenyl group, Fig. 1(a), with a distance of O to the aromatic midpoint M of only 3.111(2) Å. If the O–H bond is normalized to the standard value of 0.98 Å an H···M distance of 2.17 Å and an O–H···M angle of 160° are obtained. These are very short distances; the H···M distance actually is just about the shortest ever found in an intermolecular O–H···Ph interaction (see below). The oxygen atom is positioned close to the ring axis, and the hydrogen atom resides almost exactly over the ring midpoint. The angle between the O···M line and the ring normal vector, ω(O), is only 6.3°. Further geometrical parameters are given in Tables 2 and 3; note in particular that because of the close to centred geometry, the individual H···C and O···C distances are all significantly longer than the corresponding distances to the midpoint.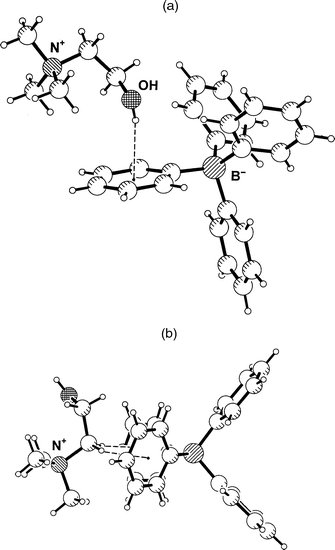 | ||
| Fig. 1 Crystal structure of choline tetraphenylborate, 1. (a) One formula unit, showing the short O–H···Ph hydrogen bond. (b) The shortest approach between the quaternary ammonium group and the Ph4B− ion, associated with a pair of C–H···Ph contacts. | ||
| H···A/Å | X···A/Å | X-H···A/° | |
|---|---|---|---|
| Compound 1 | |||
| O–H···Ph(ring 3) | 2.17 | 3.111(2) | 160 |
| Compound 2 | |||
| O1–H···Ow2 | 1.79 | 2.729(2) | 160 |
| O2–H···Ow1 | 1.67 | 2.633(2) | 166 |
| O3–H···Ow2 | 2.01 | 2.723(2) | 128 |
| N–H···O1 | 2.34 | 2.841(2) | 109 |
| N–H···O2 | 2.34 | 2.829(2) | 108 |
| N–H···O3 | 2.14 | 2.700(2) | 112 |
| Ow1–H1···O1 | 1.82 | 2.782(2) | 167 |
| Ow1–H2···Ph(ring 4) | 2.53 | 3.299(2) | 135 |
| Ow2–H1···O2 | 1.75 | 2.685(2) | 160 |
| Ow2–H2···Ph(ring 3) | 2.18 | 3.068(2) | 151 |
| Compound 3 | |||
| O1–H···O2 | 1.69 | 2.663(5) | 175 |
| N–H1···O2 | 2.16 | 3.181(3) | 169 |
| N–H3···Ow | 1.73 | 2.759(6) | 173 |
| N–H2···Ph(ring 1) | 2.48 | 3.177(6) | 124 |
| Ow–H1···Ph(ring 4) | 2.43 | 3.296(6) | 146 |
| Ow–H2···Ph(ring 2) | 2.31 | 3.229(6) | 157 |
| Compound | Typea | H···M/Å | O···M/Å | O–H···M/° | H···C/Å | O···C/Å | ω(O)/° | Donor | Acceptor | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a Inter-/intra-molecular hydrogen bond. | ||||||||||
| TITKIQ | Intra | 2.15 | 3.03 | 148 | 2.36–2.82 | 3.17–3.45 | 6.4 | C(sp3)OH (neutral) | Bz | 21 |
| 1 | Inter | 2.17 | 3.11 | 160 | 2.49–2.70 | 3.30–3.51 | 6.3 | R+C(sp3)OH | Ph4B− | — |
| 2 | Inter | 2.18 | 3.07 | 151 | 2.51–2.63 | 3.20–3.52 | 8.0 | H2O (R+ –OH co-ord.) | Ph4B− | — |
| PIPGIE | Inter | 2.18 | 3.14 | 164 | 2.47–2.72 | 3.37–3.50 | 2.9 | H2O (Na+ co-ord.) | Ph–O− | 22 |
| ZUTNEH | Inter | 2.21 | 3.14 | 158 | 2.48–2.71 | 3.40–3.47 | 1.5 | H2O (Mn+ co-ord.) | Ph4B− | 23 |
| TBATPB | Inter | 2.22 | 3.19 | 166 | 2.41–2.82 | 3.35–3.60 | 6.1 | H2O (NH+ co-ord.) | Ph4B− | 19 |
| ZOZPOT | Inter | 2.23 | 3.18 | 162 | 2.55–2.68 | 3.31–3.66 | 8.5 | H2O (Li+ co-ord.) | Ph4B− | 24 |
| GELQUJ | Intra | 2.25 | 3.23 | 172 | 2.03–3.14 | 2.84–4.08 | 33.0 | C(sp3)OH (neutral) | Bz | 25 |
| JOCDEK | Inter | 2.25 | 3.21 | 167 | 2.45–2.79 | 3.26–3.71 | 10.4 | H2O (Tc+ co-ord.) | Ph4B− | 26 |
| SEYYUQ | Inter | 2.25 | 3.09 | 143 | 2.19–2.86 | 3.17–3.60 | 10.3 | C(sp3)OH (neutral) | Ph–NH2 | 27 |
| SOCLIF | Inter | 2.27 | 3.21 | 161 | 2.32–2.98 | 3.27–3.73 | 10.8 | C(sp3)OH (neutral) | Anthryl | 28 |
| TBATPB | Inter | 2.27 | 3.12 | 144 | 2.46–2.86 | 3.40–3.44 | 0.5 | H2O (NH+ co-ord.) | Ph4B− | 19 |
The intermolecular interactions of the quaternary ammonium group are also of interest. This group is found tightly chelated between two phenyl rings of an adjacent Ph4B− anion, forming cation–π interactions as shown in Fig. 1(b).15 These interactions are associated with a pair of surprisingly short C–H···Ph contacts formed by the CH2 group attached to N+, with C···M distances of 3.49 and 3.41 Å, H···M distances of 2.42 and 2.38 Å, and C–H···M angles of 168 and 159°, respectively. This motif occurs in crystalline acetylcholine tetraphenylborate16 in very similar geometry.12
Triethanolammonium tetraphenylborate dihydrate, 2
Triethanolammonium was selected as a candidate cation for short O–H···Ph hydrogen bonds because in crystal structures it is always found in a bowl-shaped conformation in which the oxygen atoms of the three ethanol moieties converge towards the N+–H group, thereby shielding it from intermolecular hydrogen bonding. At the outer surface of the cation only the O–H groups at the rim of the bowl are available as strong hydrogen bond donors. Also the C–H groups may donate hydrogen bonds,1,17 but they are much weaker than those formed by O–H and are certainly not serious competitors.In the crystal structure of the tetraphenylborate salt, 2, this conformation of the cation is observed too, Fig. 2(a). The geometries of the three intramolecular N+–H···O hydrogen bonds are given in Table 2 (note that this represents a geometrically close to regular trifurcated or ‘four centre’18 hydrogen bond). Unexpectedly, the three hydroxy groups do not form hydrogen bonds with Ph4B− anions, but only with co-crystallized water molecules arranged around the rim of the bowl. Pairs of inversion related cations form tetrahydrated dimers, in which the N+–H vectors of the cations are antiparallel, Fig. 2(b). The rims of the two cations are not in direct contact with each other, but are connected by water bridges O–H···OW–H···O–H. This pattern is not completely regular because one of the hydroxy groups, O3–H, does not act as a hydrogen bond acceptor. Each of the water molecules uses one of its H atoms for hydrogen bonding to a cation, whereas the other OW–H group points radially outwards from the dimer axis, and donates an OW–H···Ph hydrogen bond to a neighboring anion. One of the two symmetry-independent OW–H···Ph hydrogen bonds is very short with H···M and OW···M distances of 2.18 and 3.068(2) Å, respectively (Table 2). Notably, the water molecule donating the shorter of the aromatic hydrogen bonds accepts two conventional O–H···OW hydrogen bonds, and is therefore more polarized and a stronger donor than the other one, which accepts only one O–H···OW interaction. The water molecule forming the shorter O–H···OW bond is placed almost exactly over the ring midpoint with an angle ω(O) of only 8.0°.
![Crystal structure of triethanolammonium tetraphenylborate dihydrate, 2. (a) One formula unit. (b) The tetrahydrated dication [(CHO2H4)3NH)+]2·4H2O, forming four Ow–H···Ph hydrogen bonds; H atoms bonded to C are omitted for clarity.](/image/article/2001/NJ/b004932h/b004932h-f2.gif) | ||
| Fig. 2 Crystal structure of triethanolammonium tetraphenylborate dihydrate, 2. (a) One formula unit. (b) The tetrahydrated dication [(CHO2H4)3NH)+]2·4H2O, forming four Ow–H···Ph hydrogen bonds; H atoms bonded to C are omitted for clarity. | ||
6-Ammonio-n-hexanoic acid tetraphenylborate monohydrate 3
The tetraphenylborate salt of 6-amino-n-hexanoic acid has been prepared in the hope that water would co-crystallize, possibly accept a strong hydrogen bond from the carboxylic acid group and thereby be strongly polarized, and donate a short O–H···Ph interaction. In the crystal structure of compound 3, however, a different hydrogen bond pattern is formed. The cation adopts a folded conformation with an intramolecular N+–H···O![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) C hydrogen bond, and the remaining two N+–H donors form hydrogen bonds with a co-crystallized water
molecule and a phenyl ring of the anion (geometries are given in Table 2). The carboxylic acid group forms a conventional
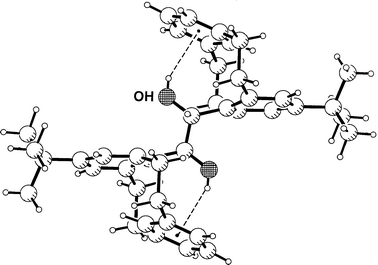
centrosymmetric dimer with a neighboring cation. The water molecule forms an interesting chelated pair of Ow–H···Ph hydrogen bonds with an anion, as is shown in Fig. 3. Such pairs of hydrogen bonds appear to be typical for tetraphenylborate salts of cations carrying bidendtate donors XH2,12 and have been found for water donors several times.9,19 The OW–H···Ph
hydrogen bond distances are H···M = 2.31 and 2.43 Å, respectively (Table 2). This is still relatively short, but unlike those in 1 and 2 not among the extreme values found in crystal structures.
C hydrogen bond, and the remaining two N+–H donors form hydrogen bonds with a co-crystallized water
molecule and a phenyl ring of the anion (geometries are given in Table 2). The carboxylic acid group forms a conventional
centrosymmetric dimer with a neighboring cation. The water molecule forms an interesting chelated pair of Ow–H···Ph hydrogen bonds with an anion, as is shown in Fig. 3. Such pairs of hydrogen bonds appear to be typical for tetraphenylborate salts of cations carrying bidendtate donors XH2,12 and have been found for water donors several times.9,19 The OW–H···Ph
hydrogen bond distances are H···M = 2.31 and 2.43 Å, respectively (Table 2). This is still relatively short, but unlike those in 1 and 2 not among the extreme values found in crystal structures.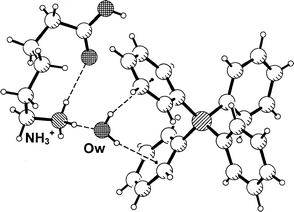 | ||
| Fig. 3 Crystal structure of 6-ammonio-n-hexanoic acid tetraphenylborate monohydrate 3; shown is one formula unit with a pair of Ow–H···Ph hydrogen bonds. | ||
Database study of short intermolecular O–H···Ph hydrogen bonds
To see how the O–H···Ph hydrogen bonds in compounds 1–3 compare with literature values, and in which kind of substances short O–H···Ph hydrogen bonds occur, a brief database survey was performed. Using the Cambridge Structural Database (CSD),20 the O–H···Ph hydrogen bonds with the ten shortest H···M distances in the current literature were identified, and their geometries listed in Table 3 (update 5.18 of the CSD, October 1999, 207507 entries, ordered and error-free organic and organometallic crystal structures with R<0.08, H-atom positions normalized). The H···M distance range of these ten interactions is 2.15–2.27 Å. The two shortest O–H···Ph interactions from the present work also fall into this range, and are added to Table 3.Of the twelve entries in Table 3, ten are from intermolecular hydrogen bonds. Of these, the two O–H···Ph bonds from compounds 1 and 2 are just about the shortest, both in terms of the H···M (2.17 Å in 1) and O···M (3.068 Å in 2) distances. The difference to the next longer examples, though, is only small. Typically, the O–H···M angles of short intermolecular O–H···Ph hydrogen bonds are found relatively linear, and angles ω(O) are small. The donor and acceptor types forming the hydrogen bonds are given in the last two columns of Table 3. Seven of the ten intermolecular bonds are with Ph4B− acceptors, confirming the particular power of this anion to accept aromatic hydrogen bonds. A different ionic acceptor in Table 3 is the phenolate ion in the crystal structure of sodium phenolate trihydrate (H···M 2.18 and O···M 3.14 Å).22
The far most frequent intermolecular donor in Table 3 is H2O, contributing seven of the ten entries. At a first sight this is surprising because water molecules by themselves are not particularly strong hydrogen bond donors, weaker than most other kinds of X–OH groups.18 On the other hand, the water molecules in Table 3 are without exception directly co-ordinated to cations, three to ammonium groups as in 3, two to alkali-metal ions (Na+ and Li+), and two to transition metal ions. Such an ion co-ordination appears to be sufficient to render water a strong enough donor to generate hydrogen bond distances H···M(Ph) around or below 2.2 Å.
Despite the dominance of ionic compounds, there are also two neutral substances contributing short intermolecular O–H···Ph bonds to Table 3. For one, (S)-1-(9-anthryl)2,2,2-trifluoroethanol, the aromatic hydrogen bonds have been discussed in detail in the original publication.28
Intramolecular hydrogen bonds must generally be treated separately from intermolecular ones. The shortest O–H···Ph bond ever found is intramolecular, with a H···M distance of 2.15 Å. It occurs in a metacyclophane21 as shown in Fig. 4. The donor as well as the acceptor of this interaction are not of strong types, and it must be suspected that the shortness of the bond is not because of a particularly strong donor–acceptor interaction, but rather because of steric compression (not unusual for intramolecular hydrogen bonds).1 Indeed, upon closer inspection it is seen that relevant parts of the molecule are substantially strained, so that the arrangement cannot directly be compared with the intermolecular hydrogen bonds in Table 3.
 | ||
| Fig. 4 The O–H···Ph hydrogen bond with the shortest H···M distance found until now (H···M 2.15, O···M 3.03 Å): crystal structure of a metacyclophane, as published by Ishi-i et al.;21 the hydrogen bond is intramolecular, and associated with steric strain. | ||
Discussion
Using the strategy to combine the acceptor Ph4B− with cationic X–OH donors, we have been able to produce very short O–H···Ph hydrogen bonds on purpose, a kind of interaction that has as yet been generated only unintendedly. The current short ends of intermolecular H···M and O···M distances are 2.17 and 3.07 Å, respectively. Such distances would in conventional O–H···O/N hydrogen bonds represent fairly long but not untypical H/O···O/N separations,18 showing that O–H···π and O–H···O/N interactions have an overlap region in their geometries.The donor and acceptor types appearing in Table 3 do not represent the strongest O–H/π(Ph) donor–acceptor combinations that one can think of. On the contrary, no particularly acidic O–H donor is present. Therefore, one may assume that O–H···Ph hydrogen bond distances can, at least in principle, be significantly reduced by bringing together stronger (i.e. more polar) OH donors and Ph acceptors in crystals. However, this is straightforward only at a first look. In fact, one must consider that strong donors tend to avoid hydrogen bonds with the relatively weak aromatic acceptors, and prefer to interact with stronger conventional hydrogen partners (like co-crystallized water molcules). This tendency will certainly sharpen with increasing donor strength. Nevertheless, this is not a fundamental chemical barrier, but more a technical obstacle that might be a challenge to overcome with crystal engineering methods.
An alternative way to reduce H···M and/or O···M distances would be to create intramolecular arrangements that are even more strained than the one shown in Fig. 4, and this might be of chemical interest as well.
Acknowledgements
AcknowledgementPart of this work was performed in the laboratory of Professor Wolfram Saenger, who is thanked for his support.References
- G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford University Press, Oxford, 1999. Search PubMed.
- A. Deeg and D. Mootz, Z. Naturforsch., Teil. B, 1993, 48, 571 Search PubMed.
- M. S. Rozenberg, T. Nishio and T. Steiner, New J. Chem., 1999, 23, 585 RSC.
- T. Steiner, E. B. Starikov, A. M. Amado and J. J. C. Teixeira-Dias, J. Chem. Soc., Perkin Trans. 2, 1995, 1312 Search PubMed; S. Harder, Chem. Eur. J., 1999, 5, 1852 CrossRef CAS.
- (a) J. F. Malone, C. M. Murray, M. H. Charlton, R. Docherty and A. J. Lavery, J. Chem. Soc., Faraday Trans., 1997, 3429 RSC; (b) D. Braga, F. Grepioni and E. Tedesco, Organometallics, 1998, 17, 2669 CrossRef CAS.
- F. H. Allen, V. J. Hoy, J. A. K. Howard, V. R. Thalladi, G. R. Desiraju, C. C. Wilson and G. J. McIntyre, J. Am. Chem. Soc., 1997, 119, 3477 CrossRef CAS.
- M. F. Perutz, G. Fermi, D. J. Abraham, C. Poyart and E. Bursaux, J. Am. Chem. Soc., 1986, 108, 1064 CrossRef CAS; G. Parkinson, A. Gunasekera, J. Vijtechovsky, X. Zhang, T. A. Kunkel, H. Berman and R. H. Ebright, Nature, Struct. Biol., 1996, 3, 837 Search PubMed; G. Xiao, S. Liu, X. Ji, W. W Johnson, J. Chen, J. F. Parsons, W. J. Stevens, G. L. Gilliland and R. N. Armstrong, Biochemistry, 1996, 35, 4753 CrossRef CAS; E. C. Dietze, C. Ibarra, M. J. Dabrowski, A. Bird and W. M. Atkins, Biochemistry, 1996, 35, 11938 CrossRef CAS.
- T. Steiner, A. M. M. Schreurs, J. A. Kanters and J. Kroon, Acta Crystallogr., Sect. D, 1998, 54, 25 CrossRef; A. Deacon, T. Gleichmann, A. J. Kalb (Gilboa), H. Price, J. Raferty, G. Bradbrook, J. Yair and J. R. Helliwell, J. Chem. Soc., Faraday Trans., 1997, 93, 4305 RSC; G. Koellner, G. Kryger, C. B. Millard, I. Silman, J. L. Sussman and T. Steiner, J. Mol. Biol., 2000, 296, 713 CrossRef CAS.
- P. K. Bakshi, A. Linden, B. R. Vincent, S. P. Roe, D. Adhikesavalu, T. S. Cameron and O. Knop, Can. J. Chem., 1994, 72, 1273 CAS.
- P. K. Bakshi, S. V. Serada, O. Knop and M. Falk, Can. J. Chem., 1994, 72, 2144 CAS.
- T. Steiner, A. M. M. Schreurs, J. A. Kanters, J. Kroon, J. van der Maas and B. Lutz, J. Mol. Struct., 1997, 436–437, 181 CrossRef.
- T. Steiner and S. A. Mason, Acta Crystallogr., Sect. B, 2000, 56, 254 CrossRef.
- S. V. Lindeman, D. Kosynkin and J. K. Kochi, J. Am. Chem. Soc., 1998, 120, 13268 CrossRef CAS.
- (a) G. M. Sheldrick, SHELXS 86, University of Göttingen, 1986; ; (b) G. M. Sheldrick, SHELXL 97, University of Göttingen, 1997..
- M. L. Verdonk, G. J. Boks, H. Kooijman, J. A. Kanters and J. Kroon, J. Comput. Aid. Mol. Des., 1993, 7, 173 Search PubMed; J. C. Ma and D. A. Dougherty, Chem. Re., 1997, 97, 1303 Search PubMed.
- N. Datta, P. Mondal and P. Pauling, Acta Crystallogr., Sect. B, 1980, 36, 906 CrossRef.
- T. Steiner, Chem. Commun., 1997, 727 RSC; G. R. Desiraju, Acc. Chem. Res., 1991, 24, 290 CrossRef CAS; G. R. Desiraju, Acc. Chem. Res., 1996, 29, 441 CrossRef CAS; T. Steiner, New J. Chem., 1998, 1099 RSC; T. Steiner and G. R. Desiraju, Chem. Commun., 1998, 891 RSC.
- G. A. Jeffrey and W. Saenger, Hydrogen Bonding in Biological Structures, Springer Verlag, Berlin, 1991; Search PubMed; G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, Oxford, 1997. Search PubMed.
- A. Aubry, J. Protas, E. Moreno-Gonzales and M. Marraud, Acta Crystallogr., Sect. B, 1977, 33, 2572 CrossRef.
- F. H. Allen and O. Kennard, Chem. Des. Autom. News, 1993, 8, 1 Search PubMed.
- T. Ishi-i, T. Sawada, S. Mataka and M. Tashiro, J. Chem. Soc., Perkin Trans. 1, 1996, 1887 RSC.
- J. Siehler, M. Pink and G. Zahn, Z. Anorg. Allg. Chem., 1994, 620, 743 CrossRef.
- P. Turner, M. J. Gunter, W. Skelton, A. H. White and T. W. Hambley, J. Chem. Res. (S), 1996, 220 Search PubMed.
- H. Schödel, T. Vaupel and H. Bock, Acta Crystallogr., Sect. C, 1996, 52, 637 CrossRef.
- S. Mataka, Y. Mitoma, T. Thiemann, M. Taniguchi and M. Tashiro, Tetrahedron, 1998, 54, 5171 CrossRef CAS.
- Y. Kani, T. Takayama, T. Sekine and H. Kudo, J. Chem. Soc., Dalton Trans., 1999, 209 RSC.
- C. N. Hodge, P. Y. S. Lam, C. J. Eyermann, P. K. Jadhav, Y. Ru, C. H. Fernandez, G. V. De Lucca, C.-H. Chang, R. F. Kaltenbach, E. R. Holler, F. Woerner, W. F. Daneker, G. Emmett, J. C. Calabrese and P. E. Aldrich, J. Am. Chem. Soc., 1998, 120, 4570 CrossRef CAS.
- H. S. Rzepa, M. L. Webb, A. M. Z. Slawin and D. J. Williams, J. Chem. Soc., Chem. Commun., 1991, 765 RSC.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2001 |