Synthesis of aromatic and heteroaromatic oligoamides on methoxypoly(ethylene glycol) as solubilizing polymer support
Burkhard König*, Ulrich Papke and Martin Rödel
Institut für Organische Chemie Universität Regensburg, D-93040, Regensburg, Germany
First published on UnassignedUnassigned4th January 2000
Abstract
A protecting-group-free procedure for the synthesis of short carbo- and heteroaromatic oligoamide chains on MeO-PEG as solubilizing support is introduced. Starting from nitrocarboxylic acids, oligoamides of carboxylic acids derived from N-methylpyrrole, N-methylimidazole and aniline are synthesized that contain up to five arene units. The polymer support facilitates work up procedures and markedly improves the otherwise poor solubility of the products. Each coupling step is monitored by 1H-NMR spectroscopy without cleavage of the product from the polymeric support.
Heteroaromatic oligoamides containing N-methylpyrrole (Py) and N-methylimidazole (Im) amino acids are an essential part of the structure of DNA-binding natural products, such as netropsin1 or distamycin.2 Inspired by these compounds and their properties, Dervan, Wemmer and others have developed synthetic heteroaromatic oligoamides that bind sequence specifically to DNA.3 The recently reported fascinating results of highly specific binding at nanomolar concentrations4 and in vivo gene regulation5 by such compounds makes their future application in gene therapy or biotechnology very likely. Baird and Dervan have provided a procedure for the synthesis of heteroaromatic oligoamides by automated solid phase synthesis, by which even extended oligoamides are available in almost every desirable combination using Boc-protected heteroaromatic amino acids as building blocks.6
We report here the synthesis of short heteroaromatic oligoamides on a soluble polymer support.7 A protecting-group-free procedure using methoxypoly(ethylene glycol) (MeO-PEG-OH), 1, with an average molecular weight of 5000 g mol−1 as an inexpensive support8 provides practical access to oligoamides for the synthesis of DNA binding agents or other purposes. Readily available nitrocarboxylic acids are used as the starting material. They are reduced to the corresponding amine after being successfully coupled to the support. It is not intended to provide a competing procedure to the reported automated solid phase synthesis6 of extended oligoamides: the well known restrictions of the MeO-PEG support in the synthesis of large peptides9 and the difficulty of automatization limit the synthesis to short oligoamides. However, synthesis on a MeO-PEG-OH solid support does offer some advantages: the handling and characterization of insoluble oligoamides is facilitated, commercially or readily available nitro compounds are employed as starting materials and every reaction step can be monitored by NMR without cleavage of the oligoamide from the polymer. This promotes the optimization of the coupling conditions for each step, which is usually required because of the significant differences in reactivity of aromatic and heteroaromatic amino acids in amide bond formation. We provide here general procedures for oligoamide synthesis on a liquid–solid support and illustrate the feasibility of the synthetic route by some examples.
Results and discussion
In the first step 1-methyl-4-nitro-1H-pyrrole-2-carbonyl chloride, 2,10 is quantitatively bound to MeO-PEG-OH (1) via an ester linkage using standard conditions (Scheme 1). For purification the polymer-bound product 3 is precipitated by addition of Et2O and collected by filtration (see general procedure 2 in the Experimental section). The excess of acid chloride is removed by washing with ether. In the next step the nitro group is reduced with HCOONH4–Pd/C in methanol solution for 1 h at room temperature (see general procedure 1 in the Experimental section). The catalyst is removed by filtration over Celite and Et2O is added to precipitate both the amine and excess HCOONH4. The solid is dissolved in dichloromethane leaving behind excess HCOONH4 and the obtained solution is used directly in the next reaction step. Reaction with 2 leads to the polymer-bound amide 4 with quantitative loading of the polymer, which is determined by NMR spectroscopy (vide infra). Repetition of the reduction-coupling cycle gives diamide 7, but the loading of the polymer decreases to 55%. A polymer loading of 83% is obtained with compound 5,10 which illustrates that the ester linkage of the heteroarene chain to MeO-PEG is not fully stable under the coupling conditions.11 Introduction of the next pyrrole yields pure triamide 9, but the loading of the support decreases to 39%. Finally, the oligoamides are cleaved from the polymer support by treatment with base, which provides 812 and 10 as carboxylic acids in good yield (see general procedure 4 in the Experimental section).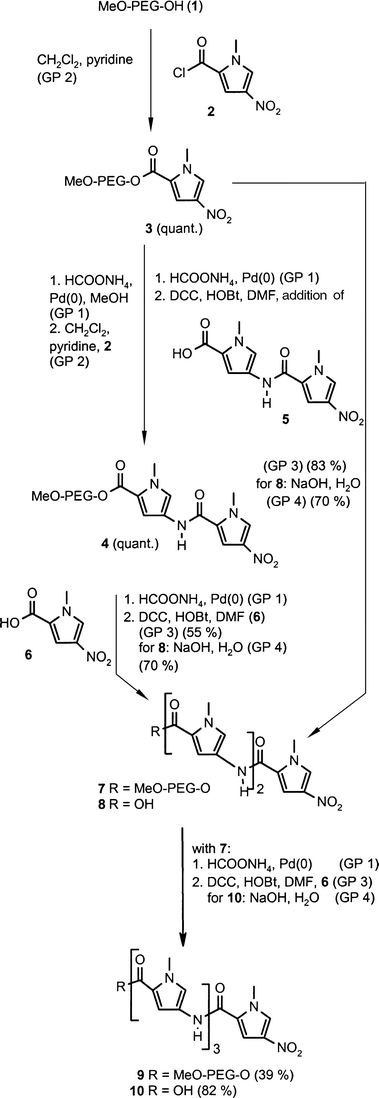 | ||
| Scheme 1 | ||
Fig. 1 shows part of the 1H-NMR spectra of 3 (top), 4 (middle) and 7 (bottom), making the growth of the oligoamide chain visible. Resonance signals of N-methyl groups successively appear, indicating the incorporation of a new pyrrole ring. Comparison of the integral of each signal with the methoxy resonance of MeO-PEG (δ=3.37), which is used as an internal standard, allows the determination of reaction yields and polymer loading.
 | ||
| Fig. 1 Part of the 1H-NMR (400 MHz, CDCl3) spectrum of polymer-bound N-methylpyrrole oligoamides. From top to bottom: compounds 3, 4 and 7. | ||
A particular advantage of oligoamide synthesis on a liquid–solid support is the possibility to control the success of coupling without cleavage from the polymer. This is illustrated by the synthesis of N-methylimidazole N-methylpyrrole diamide, 15, in Scheme 2. Though the nitrocarboxylic acid 11 can be coupled to the preceding amine, the reaction of compound 12, after reduction, with 6 is not successful. The low reactivity of the imidazole amine group in amide formation under standard conditions has been observed previously.6 To obtain 14 the use of 136 in the amide coupling is required, which was confirmed by the analysis of the polymer-bound reaction products by NMR.
 | ||
| Scheme 2 | ||
Starting from the corresponding nitrocarboxylic acids new aromatic or heteroaromatic amino acids, which have not been used for the synthesis of oligoamides so far, can be introduced. This is illustrated (Scheme 3) by the reaction of m-nitrobenzoic acid, 16-OH, which leads to the synthesis of an analog, 19, of the distamycin amide core in which the central N-methylpyrrole unit is replaced by a 1,3-disubstituted benzene ring. Again, the success of amide formation for each step is monitored by NMR. Compounds in which one amide linkage is replaced by a urea moiety, such as 23, are obtained from the reaction (Scheme 4) with nitroisocyanates (20).
 | ||
| Scheme 3 | ||
 | ||
| Scheme 4 | ||
Extended aromatic oligoamides are known for their low solubility in organic solvents.13 The unique properties of the MeO-PEG support facilitates the handling and character ization of such compounds markedly, as shown by the synthesis of 30, 31 and 35 (see Schemes 5 and 6). With nitrobenzoic acids as building blocks for oligoamide synthesis we observed a constant quantitative loading of the polymer support and complete conversion for all coupling and reduction steps up to the tetraamide 34. The growth of the aromatic oligoamide chain is monitored by the 1H-NMR resonances of the amide signals of compounds 26, 28, 32 and 34 (Fig. 2, from top to bottom).
 | ||
| Scheme 5 | ||
 | ||
| Scheme 6 | ||
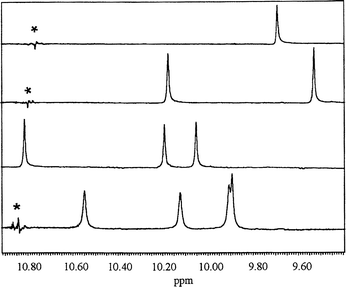 | ||
| Fig. 2 Amide proton resonance region of the 1H-NMR spectrum (400 MHz. CDCl3) of polymer-bound aromatic oligoamides. From top to bottom: compounds 26, 28, 32 and 34. * denotes a noise signal. | ||
Conclusion
In summary we have presented a simple and affordable procedure for the synthesis of short heteroaromatic and aromatic oligoamides. The described synthesis on MeO-PEG as solid support combines the convenience of solid phase procedures, such as simplified work up and removal of excess coupling reagents, with the advantages of solution reactions, such as short reaction times and the use of heterogeneous catalysts. Starting from readily available nitrocarboxylic acids, carbo- and heteroaromatic oligoamides with up to five arene units were obtained, whereby neither protecting groups, nor expensive chemicals or special apparatus are necessary. The synthesis of libraries of oligoamides from nitrocarboxylic acids may be envisaged. We hope that the reported procedure will make heteroaromatic and aromatic oligoamides more widely available and stimulate their application in biotechnology or separation techniques, such as new stationary chromatography phases.14Experimental
General
NMR spectra were recorded at 400 (1H) and 100 (13C) MHz in [D]-chloroform solutions unless otherwise stated. The multiplicity of the 13C signals was determined with the DEPT technique and are denoted as (+) for CH3 or CH, and (Cquat) for quaternary carbons. Direct inlet EI mass spectra were recorded at 70 eV on a double focusing sector field instrument (Finnigan MAT, MS8430). The source temperature was set to 200°C. High resolution experiments were performed at a resolution of 10000 (10% valley) by peak matching. In some cases where molecular ion intensities were too low for exact mass determination characteristic fragment ions (e.g., M+−CO2) were used instead. The percentage of polymer loading with oligoamide product was determined from comparison of the integrals of the 1H-NMR resonance of the terminal MeO-PEG methoxy group and a significant signal of the oligoamide residue. Py and Im indicate pyrrole and imidazole amino acids, respectively. HOBt·H2O stands for hydroxybenzotriazole and DIEA for N,N-diisopropylethylamine. mPh and pPh denote 1,3- and 1,4-benzoic amino acid, respectively.General procedure 1 (GP 1) for the reduction step
The MeO-PEG-bound nitro compound was dissolved in methanol (not more than 100 ml). For large-scale reactions a minimum amount of dichloromethane had to be added until all of the solid was dissolved. Excess HCOONH4 and 10% palladium on carbon were added, and the reaction mixture was stirred for 1 h at room temperature. The start of the reaction was indicated by the evolution of gas. In cases where this could not be observed the reaction mixture was heated with a heat gun for a short period. After 1 h at room temperature the catalyst was removed by filtration over Celite and Et2O was added (250–1000 ml depending on the scale of the reaction) to precipitate the generated MeO-PEG-bound amine and excess HCOONH4 . The solid was collected by suction and dissolved in dichloromethane leaving behind excess HCOONH4, which was filtered off. The obtained solution is directly used in GP 2 or GP 3.General procedure 2 (GP 2) for coupling with acid chlorides
To the solution obtained from GP 1 or to a solution of MeO-PEG-OH in dichloromethane, an excess of pyridine and 3–5 equiv. of the appropriate acid chloride were added and the reaction mixture was stirred for 12 h. The product was precipitated by addition of Et2O and collected by filtration. The polymer was redissolved and precipitated twice for purification, then dried in vacuo.General procedure 3 (GP 3) for the coupling step with activated acids
The appropriate acid (3–5 equiv.) was activated with DCC and HOBt·H2O (1.0 equiv. of each, corresponding to the amount of acid) in DMF (25 ml) for 4 h. To this reaction mixture the solution obtained from GP 1 was added, followed by an excess of DIEA. After 12 h stirring at room temperature the reaction mixture was filtered and the product was precipitated by addition of Et2O, and collected by filtration. The polymer was redissolved, precipitated twice and dried in vacuo.General procedure 4 (GP 4) for the cleavage of oligoamide from the polymer
The MeO-PEG-bound oligoamide was dissolved in 10 ml of aqueous 2 N NaOH and stirred at room temperature for 12 h. The solution was acidified with HCl, the precipitated oligoamide was collected by filtration and dried in vacuo.Syntheses and product characterization
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/char_e0e1.gif) =3408, 2953,
1665, 1438 cm−1. UV(CH3CN): λmax (log ε)=240 (4.36), 296
(4.43) nm. 1H-NMR (400 MHz, DMSO-d6): δ=3.82 (s, 3H),
3.86 (s, 3H), 3.96 (s, 3H), 6.85 (d, 4J=1.9 Hz, 1H), 7.05 (d,
4J=1.8 Hz, 1H), 7.26 (d, 4J=1.6 Hz, 1H), 7.42 (d, 4J=1.8
Hz, 1H), 7.59 (d, 4J=1.9 Hz, 1H), 8.18 (d, 4J=1.8 Hz, 1H),
9.94 (s, 1H), 10.28 (s, 1H), 12.16 (s, 1H). 13C-NMR (100 MHz,
DMSO-d6): δ=36.1 (+), 36.2 (+), 37.5 (+), 104.6 (+), 107.6
(+), 108.4 (+), 118.7 (+), 119.6 (Cquat), 120.3 (+), 121.5 (Cquat),
122.6 (Cquat), 122.9 (Cquat), 126.3 (Cquat), 128.3 (+), 133.8 (Cquat),
156.9 (Cquat), 158.3 (Cquat), 162.0 (Cquat). MS (EI) m/z (%)=370
(74) [M−CO2]+, 275 (100).=3427, 1701, 1648 cm−1.
UV(CH3CN): λmax (log ε)=192 (4.49) 240 (4.43), 300 (4.50), nm. 1H-NMR (400 MHz, DMSO-d6): δ=3.82 (s, 3H), 3.85 (s, 3H), 3.87 (s, 3H), 3.97 (s, 3H), 6.86 (d, 4J=1.8 Hz, 1H), 7.06 (m, 2H), 7.25 (d, 4J=1.5 Hz, 1H), 7.28 (d, 4J=1.5 Hz, 1H), 7.43 (d, 4J=1.8 Hz, 1H), 7.62 (d, 4J=1.9 Hz, 1H), 8.19 (d, 4J=1.7 Hz, 1H), 9.91 (s, 1H), 10.00 (s, 1H), 10.33 (s, 1H). 13C-NMR (100 MHz, DMSO-d6): δ=36.1 (+), 36.1 (+), 36.2 (+), 37.5 (+), 104.6 (+), 104.8 (+), 107.7 (+), 108.4 (+), 118.6
(+), 118.7 (+), 119.5 (Cquat), 120.3 (+), 121.5 (Cquat), 122.2
(Cquat), 122.6 (Cquat), 122.7 (Cquat), 123.0 (Cquat), 126.3 (Cquat), 128.2 (+), 133.8 (Cquat), 156.9 (Cquat), 158.4 (Cquat), 158.5 (Cquat), 162.0 (Cquat). MS (EI) m/z (%)=492 (100) [M−CO2]+, 397 (18), 275 (49), 153 (36). HRMS: C23H24N8O5, [M−CO2]+=492.1865±2 ppm.=3428, 1672, 1572 cm−1. UV(CH3CN): λmax (log
ε)=192 (2.36), 236 (2.21), 292 (2.20) nm. 1H-NMR (400 MHz,
DMSO-d6): δ=3.83 (s, 3H), 3.96 (s, 3H), 3.97 (s, 3H), 6.93 (d,
4J=2.0 Hz, 1H), 7.40 (d, 4J=1.9 Hz, 1H), 7.57 (s, 1H), 7.76
(d, 4J=2.0 Hz, 1H), 8.20 (d, 4J=2.0 Hz, 1H), 10.08 (s, 1H),
10.80 (s, 1H). 13C-NMR (100 MHz, DMSO-d6): δ=36.4 (+),
37.0 (+), 37.6 (+), 107.8 (+), 108.5 (+), 120.0 (Cquat), 120.6
(+), 122.0 (Cquat), 126.3 (Cquat), 126.8 (+), 128.4 (+), 134.0
(Cquat), 135.6 (Cquat), 145.3 (Cquat), 157.1 (Cquat), 159.4 (Cquat),
162.0 (Cquat). MS (EI) m/z
(%)=414 (9) [M−1]+, 399 (100).=3402, 1677, 1315 cm−1.
UV(CH3CN): λmax (log ε)=276 (4.47), 240 (4.42), 214 (4.44), 192 (4.42) nm. 1H-NMR (400 MHz, DMSO-d6): δ=3.85 (s, 3H), 3.98 (s, 3H), 6.91 (d, 4J=1.4 Hz, 1H), 7.49 (m, 2H), 7.66 (d, 3J=7.6 Hz, 1H), 7.77 (d, 4J=1.6 Hz, 1H), 7.93 (d, 3J=8.0
Hz, 1H), 8.24 (m, 2H), 10.31 (s, 1H), 10.32 (s, 1H), 12.23 (br s, 1H). 13C-NMR (100 MHz, DMSO-d6): δ=36.2 (+), 37.6 (+), 108.6 (+), 108.8 (+), 119.6 (+), 119.8 (Cquat), 120.6 (+), 122.3 (+), 122.6 (Cquat), 122.9 (+), 126.0 (Cquat), 128.7 (+), 128.8 (+), 133.9 (Cquat), 135.2 (Cquat), 138.9 (Cquat), 158.6 (Cquat), 161.9 (Cquat), 163.6 (Cquat). MS (EI) m/z (%)=411
(8) [M]+, 367 (100), 272 (84), 153 (36), 107 (20), 95 (22). HRMS: C19H17N5O6, [M]+=411.1175±2 ppm.=3408, 1676, 1547, 1314 cm−1.
UV(CH3CN): λmax (log ε)=256 (4.58), 216 (4.38), 192 (4.31)
nm. 1H-NMR (400 MHz, DMSO-d6): δ=3.81 (s, 3H), 3.91 (s,
3H), 6.65 (d, 4J=2.1 Hz, 1H), 7.15–7.25 (m, 3H), 7.29 (m, 1H),
7.73 (d, 4J=2.0 Hz, 1H), 7.93 (m, 1H), 8.22 (d, 4J=1.7 Hz,
1H), 8.30 (s, 1H), 8.66 (s, 1H), 10.09 (s, 1H). 13C-NMR (100
MHz, DMSO-d6): δ=36.1 (+), 37.6 (+), 107.7 (+), 108.5
(+), 109.9 (+), 113.4 (+), 113.5 (+), 119.1 (+), 119.8 (Cquat),
122.7 (Cquat), 126.3 (Cquat), 126.8 (+), 128.8 (+), 133.8 (Cquat),
139.0 (Cquat ), 140.4 (Cquat), 152.3 (Cquat), 158.4 (Cquat), 162.0
(Cquat). MS (FAB−): m/z (%)=425 (4) [M−1]−, 306 (100).=3315, 1645, 1526, 1323 cm−1. UV(CH3CN): λmax (log ε)=194 (4.65), 216 (4.46), 278 (4.47) nm. 1H-NMR (400 MHz, DMSO-d6): δ=7.57 (t, 3J=8.0 Hz, 1H), 7.76 (d, 3J=7.3 Hz, 1H), 7.94 (m, 4H), 8.06 (m, 1H), 8.24 (m, 2H), 8.35 (s, 1H), 8.40 (m, 2H), 10.61 (s, 1H), 10.81 (s, 1H). 13C-NMR (100 MHz, DMSO-d6): δ=119.5 (+), 120.2 (+), 123.3 (+), 123.6 (+), 125.7 (Cquat), 128.9 (+), 129.3 (+), 130.3 (+), 135.4 (Cquat), 139.0 (Cquat), 140.3 (Cquat), 143.3 (Cquat), 149.3 (Cquat), 164.1 (Cquat), 165.9 (Cquat), 167.0 (Cquat). MS (EI): m/z (%)=405 (6) [M]+, 269 (100), 150 (17), 121 (20), 104 (12), 92 (8), 76 (10). HRMS: C21H15N3O6
, [M]+=405.0955±2 ppm. Elem. anal. C21H15N3O6
: calcd. C 62.22, H 3.73, N 10.37; found C 62.01, H 3.71, N 10.07%.=3325, 1650, 1519, 1321 cm−1. UV(CH3CN): λmax (log ε)=192 (4.39), 216 (4.33), 306 (4.37) nm. 1H-NMR (400 MHz, DMSO-d6): δ=7.88 (t, 3J=8.0 Hz, 1H), 7.9–8.1 (m, 12H), 8.46 (m,
2H), 8.84 (m, 1H), 10.45 (s, 1H), 10.48 (s, 1H), 10.87 (s, 1H).
13C-NMR (100 MHz, DMSO-d6): δ=119.3 (+), 119.7 (+), 122.5 (+), 125.3 (Cquat), 126.3 (+), 128.58 (+), 128.63 (+), 129.0 (Cquat), 129.6 (Cquat), 130.1 (+), 130.2 (+), 134.2 (+), 135.9 (Cquat ), 141.9 (Cquat), 142.5 (Cquat), 143.3 (Cquat), 147.7 (Cquat), 163.6 (Cquat), 165.1 (Cquat), 165.2 (Cquat), 166.9 (Cquat). MS (EI): m/z
(%)=524 (2) [M]+, 507 (2), 480 (4), 388 (72), 286 (36), 269 (60), 150 (100), 120 (51), 104 (41), 76 (38). HRMS: C27H20N4O5
, [M−CO2]+=480.1429±5
ppm. Elem. anal. C28H20N4O7(H2O): calcd. C 61.99, H 4.09, N 10.33; found C 62.23, H 3.98, N 9.80%.3307, 1648, 1518, 1321 cm−1. UV(CH3CN): λmax (log ε)=194 (4.60), 252 (4.38) nm. 1H-NMR (400 MHz, DMSO-d6): δ=7.59 (t, 3J=7.9 Hz, 1H), 7.79 (d, 3J=7.9 Hz, 1H), 7.9–8.1 (m, 13H), 8.24 (m, 2H), 8.40 (m, 3H), 10.45 (s, 1H), 10.46 (s, 1H), 10.61 (s, 1H), 10.81 (s, 1H), 12.74 (br
s, 1H). 13C-NMR (100 MHz, DMSO-d6): δ=119.3 (+), 119.4 (+), 120.1 (+), 123.2 (+), 123.5 (+), 123.6 (+), 125.2 (Cquat), 128.6 (+), 128.8 (+), 129.0 (Cquat), 129.2 (+), 130.1 (+), 135.3 (Cquat
), 138.9 (Cquat), 140.2 (Cquat), 142.4 (Cquat), 142.5 (Cquat), 143.4 (Cquat), 149.2 (Cquat), 164.0 (Cquat), 165.1 (Cquat), 165.2 (Cquat), 165.7 (Cquat), 166.9 (Cquat). MS (EI): m/z
(%)=599 (0.1) [M−CO2]+, 507 (2), 286 (18), 269 (16), 167 (21), 150 (35), 120 (100). Elem. anal. C35H25N5O8(H2O): calcd. C 63.54, H 4.11, N 10.59; found C 63.46, H 4.04, N 10.08%.
=3408, 2953,
1665, 1438 cm−1. UV(CH3CN): λmax (log ε)=240 (4.36), 296
(4.43) nm. 1H-NMR (400 MHz, DMSO-d6): δ=3.82 (s, 3H),
3.86 (s, 3H), 3.96 (s, 3H), 6.85 (d, 4J=1.9 Hz, 1H), 7.05 (d,
4J=1.8 Hz, 1H), 7.26 (d, 4J=1.6 Hz, 1H), 7.42 (d, 4J=1.8
Hz, 1H), 7.59 (d, 4J=1.9 Hz, 1H), 8.18 (d, 4J=1.8 Hz, 1H),
9.94 (s, 1H), 10.28 (s, 1H), 12.16 (s, 1H). 13C-NMR (100 MHz,
DMSO-d6): δ=36.1 (+), 36.2 (+), 37.5 (+), 104.6 (+), 107.6
(+), 108.4 (+), 118.7 (+), 119.6 (Cquat), 120.3 (+), 121.5 (Cquat),
122.6 (Cquat), 122.9 (Cquat), 126.3 (Cquat), 128.3 (+), 133.8 (Cquat),
156.9 (Cquat), 158.3 (Cquat), 162.0 (Cquat). MS (EI) m/z (%)=370
(74) [M−CO2]+, 275 (100).=3427, 1701, 1648 cm−1.
UV(CH3CN): λmax (log ε)=192 (4.49) 240 (4.43), 300 (4.50), nm. 1H-NMR (400 MHz, DMSO-d6): δ=3.82 (s, 3H), 3.85 (s, 3H), 3.87 (s, 3H), 3.97 (s, 3H), 6.86 (d, 4J=1.8 Hz, 1H), 7.06 (m, 2H), 7.25 (d, 4J=1.5 Hz, 1H), 7.28 (d, 4J=1.5 Hz, 1H), 7.43 (d, 4J=1.8 Hz, 1H), 7.62 (d, 4J=1.9 Hz, 1H), 8.19 (d, 4J=1.7 Hz, 1H), 9.91 (s, 1H), 10.00 (s, 1H), 10.33 (s, 1H). 13C-NMR (100 MHz, DMSO-d6): δ=36.1 (+), 36.1 (+), 36.2 (+), 37.5 (+), 104.6 (+), 104.8 (+), 107.7 (+), 108.4 (+), 118.6
(+), 118.7 (+), 119.5 (Cquat), 120.3 (+), 121.5 (Cquat), 122.2
(Cquat), 122.6 (Cquat), 122.7 (Cquat), 123.0 (Cquat), 126.3 (Cquat), 128.2 (+), 133.8 (Cquat), 156.9 (Cquat), 158.4 (Cquat), 158.5 (Cquat), 162.0 (Cquat). MS (EI) m/z (%)=492 (100) [M−CO2]+, 397 (18), 275 (49), 153 (36). HRMS: C23H24N8O5, [M−CO2]+=492.1865±2 ppm.=3428, 1672, 1572 cm−1. UV(CH3CN): λmax (log
ε)=192 (2.36), 236 (2.21), 292 (2.20) nm. 1H-NMR (400 MHz,
DMSO-d6): δ=3.83 (s, 3H), 3.96 (s, 3H), 3.97 (s, 3H), 6.93 (d,
4J=2.0 Hz, 1H), 7.40 (d, 4J=1.9 Hz, 1H), 7.57 (s, 1H), 7.76
(d, 4J=2.0 Hz, 1H), 8.20 (d, 4J=2.0 Hz, 1H), 10.08 (s, 1H),
10.80 (s, 1H). 13C-NMR (100 MHz, DMSO-d6): δ=36.4 (+),
37.0 (+), 37.6 (+), 107.8 (+), 108.5 (+), 120.0 (Cquat), 120.6
(+), 122.0 (Cquat), 126.3 (Cquat), 126.8 (+), 128.4 (+), 134.0
(Cquat), 135.6 (Cquat), 145.3 (Cquat), 157.1 (Cquat), 159.4 (Cquat),
162.0 (Cquat). MS (EI) m/z
(%)=414 (9) [M−1]+, 399 (100).=3402, 1677, 1315 cm−1.
UV(CH3CN): λmax (log ε)=276 (4.47), 240 (4.42), 214 (4.44), 192 (4.42) nm. 1H-NMR (400 MHz, DMSO-d6): δ=3.85 (s, 3H), 3.98 (s, 3H), 6.91 (d, 4J=1.4 Hz, 1H), 7.49 (m, 2H), 7.66 (d, 3J=7.6 Hz, 1H), 7.77 (d, 4J=1.6 Hz, 1H), 7.93 (d, 3J=8.0
Hz, 1H), 8.24 (m, 2H), 10.31 (s, 1H), 10.32 (s, 1H), 12.23 (br s, 1H). 13C-NMR (100 MHz, DMSO-d6): δ=36.2 (+), 37.6 (+), 108.6 (+), 108.8 (+), 119.6 (+), 119.8 (Cquat), 120.6 (+), 122.3 (+), 122.6 (Cquat), 122.9 (+), 126.0 (Cquat), 128.7 (+), 128.8 (+), 133.9 (Cquat), 135.2 (Cquat), 138.9 (Cquat), 158.6 (Cquat), 161.9 (Cquat), 163.6 (Cquat). MS (EI) m/z (%)=411
(8) [M]+, 367 (100), 272 (84), 153 (36), 107 (20), 95 (22). HRMS: C19H17N5O6, [M]+=411.1175±2 ppm.=3408, 1676, 1547, 1314 cm−1.
UV(CH3CN): λmax (log ε)=256 (4.58), 216 (4.38), 192 (4.31)
nm. 1H-NMR (400 MHz, DMSO-d6): δ=3.81 (s, 3H), 3.91 (s,
3H), 6.65 (d, 4J=2.1 Hz, 1H), 7.15–7.25 (m, 3H), 7.29 (m, 1H),
7.73 (d, 4J=2.0 Hz, 1H), 7.93 (m, 1H), 8.22 (d, 4J=1.7 Hz,
1H), 8.30 (s, 1H), 8.66 (s, 1H), 10.09 (s, 1H). 13C-NMR (100
MHz, DMSO-d6): δ=36.1 (+), 37.6 (+), 107.7 (+), 108.5
(+), 109.9 (+), 113.4 (+), 113.5 (+), 119.1 (+), 119.8 (Cquat),
122.7 (Cquat), 126.3 (Cquat), 126.8 (+), 128.8 (+), 133.8 (Cquat),
139.0 (Cquat ), 140.4 (Cquat), 152.3 (Cquat), 158.4 (Cquat), 162.0
(Cquat). MS (FAB−): m/z (%)=425 (4) [M−1]−, 306 (100).=3315, 1645, 1526, 1323 cm−1. UV(CH3CN): λmax (log ε)=194 (4.65), 216 (4.46), 278 (4.47) nm. 1H-NMR (400 MHz, DMSO-d6): δ=7.57 (t, 3J=8.0 Hz, 1H), 7.76 (d, 3J=7.3 Hz, 1H), 7.94 (m, 4H), 8.06 (m, 1H), 8.24 (m, 2H), 8.35 (s, 1H), 8.40 (m, 2H), 10.61 (s, 1H), 10.81 (s, 1H). 13C-NMR (100 MHz, DMSO-d6): δ=119.5 (+), 120.2 (+), 123.3 (+), 123.6 (+), 125.7 (Cquat), 128.9 (+), 129.3 (+), 130.3 (+), 135.4 (Cquat), 139.0 (Cquat), 140.3 (Cquat), 143.3 (Cquat), 149.3 (Cquat), 164.1 (Cquat), 165.9 (Cquat), 167.0 (Cquat). MS (EI): m/z (%)=405 (6) [M]+, 269 (100), 150 (17), 121 (20), 104 (12), 92 (8), 76 (10). HRMS: C21H15N3O6
, [M]+=405.0955±2 ppm. Elem. anal. C21H15N3O6
: calcd. C 62.22, H 3.73, N 10.37; found C 62.01, H 3.71, N 10.07%.=3325, 1650, 1519, 1321 cm−1. UV(CH3CN): λmax (log ε)=192 (4.39), 216 (4.33), 306 (4.37) nm. 1H-NMR (400 MHz, DMSO-d6): δ=7.88 (t, 3J=8.0 Hz, 1H), 7.9–8.1 (m, 12H), 8.46 (m,
2H), 8.84 (m, 1H), 10.45 (s, 1H), 10.48 (s, 1H), 10.87 (s, 1H).
13C-NMR (100 MHz, DMSO-d6): δ=119.3 (+), 119.7 (+), 122.5 (+), 125.3 (Cquat), 126.3 (+), 128.58 (+), 128.63 (+), 129.0 (Cquat), 129.6 (Cquat), 130.1 (+), 130.2 (+), 134.2 (+), 135.9 (Cquat ), 141.9 (Cquat), 142.5 (Cquat), 143.3 (Cquat), 147.7 (Cquat), 163.6 (Cquat), 165.1 (Cquat), 165.2 (Cquat), 166.9 (Cquat). MS (EI): m/z
(%)=524 (2) [M]+, 507 (2), 480 (4), 388 (72), 286 (36), 269 (60), 150 (100), 120 (51), 104 (41), 76 (38). HRMS: C27H20N4O5
, [M−CO2]+=480.1429±5
ppm. Elem. anal. C28H20N4O7(H2O): calcd. C 61.99, H 4.09, N 10.33; found C 62.23, H 3.98, N 9.80%.3307, 1648, 1518, 1321 cm−1. UV(CH3CN): λmax (log ε)=194 (4.60), 252 (4.38) nm. 1H-NMR (400 MHz, DMSO-d6): δ=7.59 (t, 3J=7.9 Hz, 1H), 7.79 (d, 3J=7.9 Hz, 1H), 7.9–8.1 (m, 13H), 8.24 (m, 2H), 8.40 (m, 3H), 10.45 (s, 1H), 10.46 (s, 1H), 10.61 (s, 1H), 10.81 (s, 1H), 12.74 (br
s, 1H). 13C-NMR (100 MHz, DMSO-d6): δ=119.3 (+), 119.4 (+), 120.1 (+), 123.2 (+), 123.5 (+), 123.6 (+), 125.2 (Cquat), 128.6 (+), 128.8 (+), 129.0 (Cquat), 129.2 (+), 130.1 (+), 135.3 (Cquat
), 138.9 (Cquat), 140.2 (Cquat), 142.4 (Cquat), 142.5 (Cquat), 143.4 (Cquat), 149.2 (Cquat), 164.0 (Cquat), 165.1 (Cquat), 165.2 (Cquat), 165.7 (Cquat), 166.9 (Cquat). MS (EI): m/z
(%)=599 (0.1) [M−CO2]+, 507 (2), 286 (18), 269 (16), 167 (21), 150 (35), 120 (100). Elem. anal. C35H25N5O8(H2O): calcd. C 63.54, H 4.11, N 10.59; found C 63.46, H 4.04, N 10.08%.Acknowledgements
This work was supported by the Fonds der Chemischen Industrie. M.R. thanks the Land Niedersachsen, Germany, for a graduate fellowship.References
- A. C. Finlay, F. A. Hochstein, B. A. Sobin and F. X. Murphy, J. Am. Chem. Soc., 1951, 73, 341 CrossRef CAS.
- F. Arcamone, S. Penco, O. Orezzi, V. Nicolella and A. Pirelli, Nature (London), 1964, 203, 1064 CAS.
- M. Mrksich, W. S. Wade, T. J. Dwyer, B. H. Geierstanger, D. E. Wemmer and P. B. Dervan, Proc. Natl. Acad. Sci. U.S.A., 1992, 89, 7586 Search PubMed; D. S. Johnson and D. L. Boger, in Comprehensive Supramolecular Chemistry, eds. J. Atwood, J. E. D. Davies, D. D. Macnicol, F. Vögtle, J.-M. Lehn and Y. Murakami, Pergamon Press, Oxford, 1996, vol. 4, p. 73 and cited references. Search PubMed; P. E. Nielsen, Chem. Eur. J., 1997, 3, 505 and cited references. Search PubMed; G. Jia, H. Iida and J. W. Lown, Chem. Commun., 1999, 119. Search PubMed.
- J. W. Trauger, E. E. Baird and P. B. Dervan, Nature (London), 1996, 382, 559 CrossRef CAS; J. M. Turner, E. E. Baird and P. B. Dervan, J. Am. Chem. Soc., 1997, 119, 7636 CrossRef CAS; S. E. Swalley, E. E. Baird and P. B. Dervan, Chem. Eur. J., 1997, 3, 1600 CAS; S. E. Swalley, E. E. Baird and P. B. Dervan, J. Am. Chem. Soc., 1997, 119, 6953 CrossRef CAS.
- J. M. Gottesfield, L. Nealy, J. W. Trauger, E. E. Baird and P. B. Dervan, Nature (London), 1997, 387, 202 CrossRef CAS.
- E. E. Baird and P. B. Dervan, J. Am. Chem. Soc., 1996, 118, 6141 CrossRef CAS.
- B. König and M. Rödel, Chem. Commun., 1998, 605 RSC.
- For the use of polyethylene glycol as a support in synthesis, see: M. Mutter, H. Hagenmaier and E. Bayer, Angew. Chem., 1971, 83, 883; Search PubMed; Angew. Chem., Int. Ed. Engl., 1971, 10, 811; Search PubMed; R. N. V. Pillai and M. Mutter, Top. Curr. Chem., 1982, 106, 119; ; combinatorial libraries: H. Han, M. M. Wolfe, S. Brenner and K. D. Janda, Proc. Natl. Acad. Sci. U.S.A., 1995, 92, 6419. Search PubMed.
- D. J. Gravert and K. D. Janda, Chem. Rev., 1997, 97, 489 CrossRef CAS.
- J. W. Lown and K. Krowicki, J. Org. Chem., 1985, 50, 3774 CrossRef CAS.
- This limitation of PEG supports has been described for other peptide syntheses earlier. For examples, see ref. 9..
- J. S. Taylor, P. G. Schultz and P. B. Dervan, Tetrahedron, 1984, 40, 457 CrossRef CAS; M. Bialer, B. Yagen and R. Mechoulam, Tetrahedron, 1978, 34, 2389 CrossRef CAS.
- H. Bredereck and H. von Schuh, Chem. Ber., 1948, 81, 215 Search PubMed; S. Gould and D. A. Laufer, J. Magn. Reson., 1979, 34, 37 CAS.
- B. König, H. Buchholz, M. Rödel and M. Schulze, DE Pat. 198 05 431.9, 1998; European, Japanese and US patents pending. Search PubMed.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2000 |