Tubular electrolytic hydride generator for continuous and flow injection sample introduction in atomic absorption spectrometry
Francisco Laborda*, Eduardo Bolea and Juan R. Castillo
Analytical Spectroscopy and Sensors Group, Department of Analytical Chemistry, University of Zaragoza, 50009, Zaragoza, Spain
First published on UnassignedUnassigned7th January 2000
Abstract
An electrolytic hydride generation system for sample introduction in atomic spectrometry is described. The system uses a tubular electrolytic flow-through cell with a concentric arrangement and a packed cathode made of vitreous carbon. The large cathode surface area (about 120 cm2) gives efficiencies in hydride production of >90% for As(III), Se(IV) and Sb(III). Samples can be introduced in both continuous and flow injection modes, because the low dead volume (390 µl) of the generator complies with the dispersion requirements of flow injection systems. The generator was coupled to a flame heated quartz tube for atomic absorption measurements. The detection limits for As, Se and Sb in aqueous solutions were 5.2, 2.5 and 1.4 ng ml−1, respectively, with relative standard deviations of 0.7–3.3%.
The generation of volatile hydrides of a number of elements (As, Bi, Ge, In, Pb, Sn, Sb, Se, Te and Tl) is a well established sample introduction technique in atomic spectrometry when low detection limits have to be achieved.1 Advantages center on the higher transport efficiency of the analyte, close to 100%, compared to solution nebulization (1–10% for pneumatic nebulizers). The technique is not free from interferences, the transition and noble metals especially reduce or suppress the production of hydrides, depending on the experimental conditions.2
Hydride generation is usually performed using sodium tetrahydroborate in acidic media. The nascent hydrogen produced reacts with the analyte and its hydride is formed. In spite of its widespread use, sodium tetrahydroborate has several disadvantages. The reagent is expensive and may be a source of contamination. In addition, its solutions are unstable and have to be prepared daily.
Electrolytic hydride generation is an alternative to the chemical generation based on the sodium tetrahydroborate–acid system, because fewer reagents are required and experimental conditions are less critical.3 Additional advantages include the reduction of interferences from transition metals4–7 and the reduced influence of the oxidation states of the analyte on the hydride yield,3,8 depending on the type of cathode material used. In electrolytic hydride generation, the hydride is formed in the cathodic space of an electrolytic cell. The process is considered to take place in at least three sequential events:3,8,9 reduction and deposition of the analyte onto the surface of the cathode (or at specific surface sites), stepwise reaction of the deposited metal with nascent hydrogen generated on the cathode by reduction of protons, and desorption of the hydride. Simultaneously, water is oxidized in the separated anodic space and oxygen is formed.
The determination of arsenic based on the electrolytic generation of its hydride was first reported by Bloxam in 1861.10,11 The method was applied by Grant to the determination of antimony in 1928.12 In the twenties, Paneth et al. generated tin13,14 and germanium hydrides electrolytically.15 Analytical applications of the electrolytic hydride generation were also reported by Rigin et al. from 1966, who described the determination of arsenic16–18 and tin19 by atomic absorption and atomic fluorescence based on the electrolytic production of their hydrides in batch generators. More recent reports3–9,20,21 employ continuous and flow injection generators, which are easily automated and need smaller sample volumes. The generators most often used in these works are flow-through cells of the thin-layer type,3–9,20 where electrodes are made of sheets of a suitable material (platinum, lead, vitreous carbon, pyrolitic graphite) and the cathodic and anodic spaces are separated by an ion exchange membrane or porous glass frits. Hueber and Winefordner20 described a more complex design that used a coil and a mesh of wire as the cathode and anode, respectively. The generator briefly described by Schaummlöffel and Neidhart21 used a packed cathode of fibrous carbon. Using these systems, electrolytic generation of hydrides of arsenic, selenium and antimony has been reported. Generation efficiencies from 30 up to 98% have been obtained depending on the electrode material and cell design.
To achieve a high generation efficiency in a short period of time, an electrolytic generator should ensure a high mass transfer rate of the analyte to the cathode surface. Using flow-through cells, this can be achieved with cathodes having large surface areas such as porous cathodes or by using thin-layer designs having a large electrode surface area and low solution thickness. In addition, if flow injection systems are going to be used, the cell should possess a low void volume to avoid signal broadening. Reticulated vitreous carbon (RVC) has been used extensively in electrochemical flow-through cells22 because of its chemical inertness, hydrodynamic and electrochemical properties, and its low cost. On the other hand, carbon has a medium hydrogen overvoltage, which ensures a better generation efficiency than using platinum.
The aim of this work is to evaluate a new design of electrolytic hydride generator for sample introduction in atomic spectrometry based on the use of a porous cathode made of reticulated vitreous carbon and a concentric arrangement of the flow-through electrolytic cell.
Experimental
Instrumentation
A Perkin Elmer (Norwalk, CT, USA) Model 2380 atomic absorption spectrometer, equipped with a flame heated quartz tube atomizer (168 mm length, 12 mm id) was used for atomic absorption measurements. Hollow cathode lamps of arsenic, selenium and antimony were used; operating conditions are listed in Table 1. Flow injection signals were evaluated by their peak heights.| Parameter | As | Se | Sb |
|---|---|---|---|
| Hollow cathode lamp/mA | 25 | 16 | 20 |
| Wavelength/nm | 193.7 | 196.0 | 217.6 |
| Slit width/nm | 0.7 | 2.0 | 0.2 |
| Electrolyte solution/mol H2SO4 l−1 | 0.5 | 0.5 | 0.5 |
| Electrolyte flow rate/ml min−1 | 5.4 | 5.4 | 5.4 |
| Current density/mA cm−2 | 8.3 | 8.3 | 8.3 |
| Nitrogen flow rate/l min−1 | 1.0 | 1.0 | 1.0 |
Generator design
The generator consists of a tubular electrolytic flow-through cell, where the cathode and anode spaces have a concentric configuration. Fig. 1 shows a cross-section view of the hydride generator. It consists of a central cathode made of reticulated vitreous carbon (RVC) or crushed reticulated vitreous carbon (CRVC) inside a porous ceramic tube (7 mm id, 15 mm length of cathode) and an anode made of a coil of platinum wire (30 cm length, 0.25 mm diameter) surrounding the ceramic tube. The cathode and anode are placed inside of a polytetrafluoroethylene (PTFE) tube which serves as the anodic space. Both ends are closed with PTFE pieces sealed with an epoxy resin. The cathodic and anodic spaces are provided with solution inlets and outlets and external electrical contacts. | ||
| Fig. 1 Cross-section view of the electrolytic flow-through cell. (1) Porous ceramic tube, (2) graphite electric contact, (3) CRVC or RVC cathode, (4) platinum wire anode. | ||
The dc power supply used was a Promax FAC-662B, operated in the constant current mode.
Reagents and standard solutions
A standard solution of Se(IV) was prepared by dissolving SeO2 (Merck, Elmsford, NY, USA) in ultrapure water. A standard solution of Se(VI) was prepared by dissolving Na2SeO4 (Sigma) in ultrapure water. A standard solution of As(III) was prepared by dissolving As2O3 (Merck) in a small volume of 20% KOH solution, neutralizing with H2SO4 and diluting with ultrapure water. A standard solution of As(V) was prepared by dissolving Na2HAsO4 (Panreac, Barcelona, Spain) in ultrapure water. Antimony potassium tartrate (Merck) was dissolved in ultrapure water to prepare a standard solution of Sb(III). A standard solution of Sb(V) was prepared by dissolving Sb2O5 (Merck) in a small volume of concentrated HCl, and diluting with ultrapure water. Working standard solutions were prepared by further dilution in 0.5 mol l−1 H2SO4, except where indicated otherwise. Solutions containing Sb(III) were made up to 1% (w/v) with L-cysteine to avoid oxidation of the analyte.
Hydride generation
Electrochemical hydride generation was accomplished in continuous or flow injection mode by using the system shown in Fig. 2. The complete system consisted of a peristaltic pump (Gilson Minipuls III, Lilliers le Bel, France), a six ways injection valve (Omnifit, Cambridge, UK) and a gas–liquid separator with forced outlet, described previously.23 Nitrogen (99.998%) was used as the carrier gas, being introduced into the gas–liquid separator and controlled by a flow meter. PTFE tubing (id 0.8 mm) was used for all connections.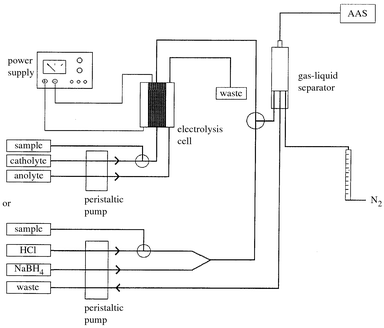 | ||
| Fig. 2 Diagram of the electrolytic and chemical hydride generation systems coupled to the flame heated quartz tube AAS. | ||
In continuous mode, solutions of the analyte acidified with H2SO4 or HCl (0.5 mol l−1) were used as the catholyte and solutions of H2SO4 or HCl (20% hydroxylamine hydrochloride) of the same concentration were used as the anolyte. The catholyte and anolyte were continuously pumped at a flow rate of 5.4 ml min−1 and a voltage (10–15 V) was applied to deliver a constant current (1 A). Oxygen was produced at the anode, whereas the analyte hydride and hydrogen were formed at the cathode. The catholyte and reaction products were delivered to the gas–liquid separator from which the gases were directed to the atomizer by a nitrogen stream at a flow rate of 1.0 l min−1. In the flow injection mode, a solution of H2SO4 or HCl was used as catholyte and a volume of 420 µl of analyte acidified with the same acid was injected into the catholyte stream.
For chemical hydride generation (Fig. 2) sodium tetrahydroborate (flow rate: 1.0 ml min−1) and HCl (2.5 ml min−1) were continously pumped through a T-piece. The electrolytic hydride generator was exchanged by a reaction coil (length 3.5 cm) and this stream was conducted to the gas–liquid separator as described above.
Results and discussion
Electrochemical hydride generator design
The proposed generator was designed for compactness, large cathode surface area, low dead volume, the use of non expensive materials and to maintain or improve the analytical performance of chemical generation.In contrast to thin-layer designs,3–9,20 a concentric arrangement of the cathodic and anodic compartments was selected. This arrangement implies the use of a flow-through cathode made of a porous or a packed particulated material and allows a high surface to volume ratio. In this context, the cathode area is related to the efficiency of the hydride generation and the volume of the cathodic compartment to the dispersion of the injected analyte by the void volume of the cell. Although RVC was initially selected as the cathode material, a packed cathode made of crushed RVC was preferred, due to its higher specific area and better sensitivity (34% improvement) obtained with the same volume of cathode. On the other hand, the volume of the cathode was reduced to 0.58 cm3 (1.5 cm length) without loss of sensitivity. The void volume was reduced to 0.39 cm3, which means a porosity of the packed cathode of about 70%. Considering that particles of crushed RVC are spherical and of average diameter 200 µm, the cathode active surface is around 120 cm2, much higher than in thin-layer generators where typical active surfaces are 10 cm2. These generators are working at current densities from 150–2003,8 to 900 mA cm−2,7 whereas the proposed generator works at a lower current density (8.3 mA cm−2) without loss of efficiency in hydride production.
Study of operating conditions
In the first instance, operating conditions were studied by the introduction of the analyte in a continuous mode. These optimized conditions were confirmed by flow injection and are summarized in Table 1. | ||
| Fig. 3 Effect of the electrolytic current on the analytical signal of Se(IV): continuous sample introduction; electrolyte concentration, H2SO4 0.5 mol l−1; sample flow rate, 5.4 ml min−1; 100 ng ml−1 Se. | ||
The circulating current is limited by the applied voltage and the ohmic resistance of the electrolytic cell. In our design, the main contribution to this resistance arose from the electrolyte and its concentration. The effect of electrolyte concentration on sensitivity was studied in the range of 0.01–2.0 mol l−1 for H2SO4, maintaining a constant current of 1 A. Results presented in Fig. 4 for antimony show that the concentration of the electrolyte is not critical for electrolytic hydride generation at this current; similar conclusions were obtained for the three elements studied. On the other hand, as the electrolyte concentration was reduced, the voltage applied to obtain the current of 1 A had to be increased due to the lower conductivity of the solution.
 | ||
| Fig. 4 Effect of the electrolyte concentration on the analytical signal of Sb(III): (A) H2SO4, (B) HCl; continuous sample introduction; sample flow rate, 5.4 ml min−1; 100 ng ml−1 Sb. | ||
Hydrochloric acid was also tested as the electrolyte, although this acid offered lower sensitivity, as it is shown in Fig. 4. In order to avoid the oxidation of chloride to chlorine, 20% hydroxylamine hydrochloride was added to the anolyte.3,8 Sulfuric acid 0.5 mol l−1 was finally chosen as the electrolyte for further studies, because lower electrolytic concentration produced an overheating of the flowing solutions due to the higher applied voltages.
 | ||
| Fig. 5 Effect of the sample flow rate on the analytical signal (A) and generation efficiency (B) of Se(IV): continuous sample introduction; electrolyte concentration, H2SO4 0.5 mol l−1; 100 ng ml−1 Se. | ||
Generation efficiency and effect of the analyte oxidation state
The efficiency of the hydride generation was estimated by measuring the fraction of analyte not volatilized and remaining in the solution during hydride generation. A solution containing 1 µg ml−1 of analyte was continuously pumped through the cathode, the generator was operated under the selected experimental conditions and the solution drained in the gas–liquid separator collected. The analyte concentration of the collected solution was determined by electrolytic hydride generation and related to its initial concentration. Due to the porosity of the ceramic tube that separates the anodic and cathodic spaces, a flow of solution from the anode to the cathode was observed while the generator was working, thus producing a dilution of the catholyte. The efficiency of hydride generation was estimated as: | (1) |
Efficiencies higher than 90% were obtained for all the elements studied in their lower oxidation states, whereas the highest oxidation states showed different behaviours. The estimated hydride generation efficiency from Se(IV) was 95%, whereas from Se(VI) was <1%. Arsine generation could be accomplished from both arsenic oxidation states although with different efficiencies, 93% from As(III) and 47% from As(V). The efficiency from Sb(III) and (V) was 92% and 40%, respectively.
Figures of merit
Analytical figures of merit for continuous sample introduction are summarized in Table 2. Detection limits in the range of 1–5 ng ml−1 and relative standard deviations of 1–4% were obtained for all the elements studied.| Analyte | Characteristic concentration/ng ml−1 | Detection limita/ng ml−1 | RSDb (%) |
|---|---|---|---|
| a3σ criteria. bn=10. | |||
| As(III) | 5.2 | 4.9 | 3.3 |
| Sb(III) | 1.4 | 0.9 | 0.7 |
| Se(IV) | 2.5 | 1.4 | 2.4 |
A comparison of continuous sample introduction between the conventional hydride generation based on reduction by tetrahydroborate and the electrolytic hydride generation showed no significant differences in analytical performance. The characteristic concentration and detection limit for selenium by using tetrahydroborate under optimum conditions were 2.8 and 1.5 ng ml−1, respectively.
When sample introduction was performed by flow injection, the void volume of the hydride generator (390 µl) contributed to the increase of the dispersion of the sample. Thus, a lower sensitivity was obtained due to the decrease in peak height of the transient signal. Selenium analytical figures of merit for flow injection sample introduction are listed in Table 3 and tetrahydroborate and electrolytic hydride generation are compared. In spite of the lower dispersion of the tetrahydroborate flow injection system, the performance of both systems was similar.
| Hydride generation | Characteristic concentration/ng ml−1 | Characteristic mass/ng | Detection limita/ng ml−1 | Db |
|---|---|---|---|---|
| a3σ criteria. bFIA dispersion: absorbance (continuous)/absorbance (peak height). | ||||
| Tetrahydroborate | 3.1 | 1.3 | 1.7 | 1.13 |
| Electrochemical | 4.1 | 1.7 | 1.9 | 1.65 |
Determination of selenium and antimony in homeopathic dilutions
Two homeopathic tinctures, Natrium Selenicosum 5 DH containing 4.56 mg l−1 of selenium as sodium selenite and Antimonium Tartaricum 5 DH containing 3.65 mg l−1 of antimony as antimony potassium tartrate, were diluted (2∶100) and analyzed according to the proposed technique. Selenium and antimony contents of 4.39 ± 0.17 mg l−1 and 3.42 ± 0.07 mg l−1 were obtained from direct calibration against aqueous standards, respectively. Results were in agreement with the contents listed by the manufacturer.Conclusions
The tubular design of the electrolytic hydride generator and the use of a packed cathode provides a large cathode surface area in a small volume. This arrangement implies an easy access of the analyte to the cathode surface which means a high hydride generation efficiency. On the other hand, the low void volume produces a small dispersion when the generator is used in a flow injection system. The vitreous carbon used as the cathode material is a good choice for the hydride generation of arsenic, selenium and antimony and the same set of experimental conditions can be used. The higher oxidation states of arsenic, antimony and selenium show a behaviour similar to that observed when tetrahydroborate is used. The hydride is not generated from Se(VI) and with lower efficiency from As(V) and Sb(V).Acknowledgements
This work has been sponsored by the DGICYT of the Spanish Ministry of Education and Science, project no. PB 97-0995.References
- J. DedinaD. L. Tsalev, Hydride Generation Atomic Absorption Spectrometry, J. Wiley & Sons, Chichester, 1995. Search PubMed.
- A. E. Smith, Analyst, 1975, 100, 300 RSC.
- W. W. Ding and R. E. Sturgeon, J. Anal. At. Spectrom., 1996, 11, 225 RSC.
- Y. Lin, X. Wang, D. Yuan, P. Yang and B. Huang, J. Anal. At. Spectrom., 1992, 7, 287 RSC.
- A. Brockmann, C. Nonh and A. Golloch, J. Anal. At. Spectrom., 1993, 8, 397 RSC.
- C. Schickling, J. Yang and J. A. C. Broekaert, J. Anal. At. Spectrom., 1996, 11, 739 RSC.
- L. F. R. Machado, A. O. Jacintho, A. A. Menegário, E. A. G. Zagatto and M. F. Giné, J. Anal. At. Spectrom., 1998, 13, 1343 RSC.
- W. W. Ding and R. E. Sturgeon, Spectrochim. Acta, Part B, 1996, 51, 1325 CrossRef.
- W. W. Ding and R. E. Sturgeon, J. Anal. At. Spectrom., 1996, 11, 421 RSC.
- C. L. Bloxam, Quart. J. Chem. Soc., 1861, 13, 12 Search PubMed.
- C. L. Bloxam, Quart. J. Chem. Soc., 1861, 13, 339 Search PubMed.
- J. Grant, Analyst, 1928, 53, 626 RSC.
- F. Paneth, Z. Elektrochem., 1923, 29, 97 Search PubMed.
- F. Paneth and E. Rabinowitsch, Ber. Dtsch. Chem. Ges., 1924, 57B, 1877 Search PubMed.
- F. Paneth and E. Rabinowitsch, Ber. Dtsch. Chem. Ges., 1925, 58B, 1138 Search PubMed.
- V. I. Rigin and N. N. Melnichenko, Zavod. Lab., 1966, 32, 394 Search PubMed.
- V. I. Rigin and G. N. Verkhoturov, Zh. Anal. Khim., 1977, 32, 1965 Search PubMed.
- V. I. Rigin, Zh. Anal. Khim., 1978, 33, 1966 Search PubMed.
- V. I. Rigin, Zh. Anal. Khim., 1979, 34, 1569 Search PubMed.
- D. M. Hueber and J. D. Winefordner, Anal. Chim. Acta, 1995, 316, 129 CrossRef CAS.
- D. Schaumlöffel and B. Neidhart, Fresenius' J. Anal. Chem., 1996, 354, 866 CAS.
- J. Wang, Electrochim. Acta, 1981, 26, 1721 CrossRef CAS.
- F. Laborda, M. T. Gómez, M. S. Jiménez, J. M. Mir and J. R. Castillo, J. Anal. At. Spectrom., 1996, 11, 1121 RSC.
| This journal is © The Royal Society of Chemistry 2000 |