The effect of oxidation and acidification on the speciation of heavy metals in sulfide-rich freshwater sediments using a sequential extraction procedure
Susan E. J.
Buykx
a,
Margriet
Bleijenberg
a,
Marc A. G. T.
van den Hoop
*a and
J. P.
Gustav Loch
b
aNational Institute of Public Health and the Environment, P.O. Box 1, 3720 BA, Bilthoven, The Netherlands. E-mail: magt.van.den.hoop@rivm.nl; Fax: +31 30 274 44 55;; Tel: Tel: +31 30 274 34 37
bDepartment of Geochemistry, Institute of Earth Sciences, Utrecht University, P.O. Box 80021, 3508 TA, Utrecht, The Netherlands
First published on 28th January 2000
Abstract
The speciation of metals in a contaminated, anoxic, sulfide-rich, freshwater sediment was determined experimentally, using a sequential extraction procedure based on the method of Tessier et al. Taking into account the advantages and disadvantages of sequential extractions, the applied methodology allowed the investigation of the influence of aeration and acidification on the distribution of various metals in the sediment. Aeration caused Zn and Cd to be released from sulfides. Carbonates were partly dissolved by the oxidation process, causing mobilisation of Ca. Fe became less mobile owing to a stronger binding to organic matter. The speciation of K, Al, Ni, Pb and Mn and to a lesser extent of Cu was not affected by aeration. As a result of acidification of the aerated sediment, Ca, Mn, Ni, Zn and Cd became more mobile owing to the dissolution of carbonates.
Introduction
Sediments consist of a complex mixture of organic and inorganic components. In the solution phase, metals are present as free ions and as inorganic and organic complexes. In the solid phase, the main adsorbing phases are clay minerals, (coatings of) Fe, Mn and Al (hydr)oxides, organic matter and, in some cases, minerals such as calcite. Metals can also be present in the lattice structure of minerals. In addition, metals can form precipitates such as metal carbonates or metal sulfides. The latter are formed in anoxic sediments, where bacteria can reduce sulfate to sulfide. The sulfide can then react with iron and trace metals to form metal sulfide precipitates. From a thermodynamic point of view, a trace metal sulfide will preferentially be formed instead of iron sulfide because of the lower stability constants.1The total trace metal content in the sediment provides important information about the pollution level if the background or geochemical composition is known. However, it is the speciation of the element that determines its physico-chemical behaviour and hence plays a key role with respect to toxicity, reactivity, mobility and bioavailability.
Direct measurement of trace elements associated with a given sediment phase is seldom feasible, owing to the complexity of the system and low concentrations of the trace elements involved. Therefore, indirect methods such as microprobe, thermodynamic models and sequential extraction techniques are used. Microprobe analysis gives quantitative information about trace metals on selected minerals or sediment particles, but a large number of individual analyses of sediment particles and mineral fragments must be performed to give a reliable “picture" of the whole sediment.2 The main problem with thermodynamic models is related to the lack of thermodynamic data and the uncertainty in these data. A sequential extraction procedure can be used to determine operationally the trace metal speciation in natural sediments, and to estimate qualitatively the metal reactivity and mobility, by comparing the speciation in sediments under different relevant environmental conditions such as redox potential or pH.3
In a sequential extraction procedure, chemical extractants of various types are successively applied to the sediment sample, each follow-up treatment being more drastic in chemical action than the previous one. The sequential extraction procedures described in the literature differ mainly in the extractants used, the order of extractions and operational details such as the solid-to-extractant ratio and extraction time.4 The results obtained by a sequential extraction procedure are operationally defined. In other words, “what is measured is defined by what is measured".5 Owing to its operational character, the results obtained by a sequential extraction procedure cannot be attributed to a specific particular solid phase.6 Therefore, results must be interpreted rather qualitatively instead of quantitatively and thus cannot be used as input data for thermodynamic equilibrium models. Sequential extraction procedures are practical, easy to perform, consider more than one phase and are used in many studies. Overall, it can be stated that the use of a sequential extraction procedure is a practical approach with several limitations. If it is used with care and the limitations are taken into account, it can be useful in the study of sediment geochemistry, particularly in those cases where the effects of one alteration (e.g., oxidation state) are examined.
The most widely used sequential extraction procedure is that proposed by Tessier et al.7 This extraction procedure results in five fractions. These fractions, although operationally defined, have been attributed to a certain sediment phase and named correspondingly. The sediment phases can be affected by short-term natural or anthropogenic changes in environmental conditions. Fraction 1 is obtained using MgCl2 and contains correspondingly the exchangeable metals. Changes in ionic composition, e.g., in estuarine waters, can alter the adsorption and desorption of metals on sediment constituents. Fraction 2, extracted with an acetate buffer, consists of metals bound to carbonates, which can be affected by the production or consumption of protons. These can be caused, for example, by anthropogenic acid deposition, natural acid production or oxidation. Fraction 3 is obtained using NH2OH·HCl and contains the metals bound to Fe and Mn (hydr)oxides, whereas fraction 4, obtained by oxidation with peroxide and extraction with NH4OAc, contains the metals bound to oxidizable compounds: organic matter and sulfides. The association of metals with these two fractions can be changed by variations in redox conditions, Fe and Mn oxides being affected by anoxic conditions and organic matter and sulfides by oxic conditions. These latter can be caused, for example, by dredging of sediments or resuspension due to storm. In oxidative conditions, sulfide will be oxidised to sulfate, releasing protons and metals. The released metals may in turn quickly be scavenged by solid phases or form complexes. Fraction 5 contains the metals in the lattice of primary and secondary minerals (residual fraction). Changes in natural environmental conditions have no effect on the release of metals in this fraction on the time-scale of a decade.
In the present study, the sequential extraction procedure developed by Tessier et al.7 was used with some modifications. First, Tessier et al.7 used dry sediment for the extraction. However, drying causes oxidation of the sediment, which can change the original speciation of the metals. In our study, wet sediment was used, in order to be able to preserve the anoxic conditions in the sediment. Second, part of the sediment was aerated and/or acidified to study the effect of the change in oxidation or protons on the distribution of the metals in the sediment. Other modifications of the procedure included an additional extraction step, fraction 0, which extracts the water-soluble metals. Fraction 5, the residual fraction, was calculated from the difference between the “total" metal content determined by aqua regia digestion of the original sediment and the sum of fractions 0–4, instead of digestion of the residual remaining after step 4 with HF and HClO4. In the literature, metal concentrations obtained by aqua regia and HF–HClO4 digestion were found to be in good agreement (average differences <7% for Cd, <10% for Cu, <6% for Ni, <12% for Zn and <13% for Mn, except for Pb, which showed average differences <37%).8,9
The aim of this work was to determine experimentally the metal speciation in anoxic, sulfide-rich, freshwater sediment and to study the influence of aeration and acidification on the distribution of the metals in the sediment.
Experimental
Sampling and sample preparation
Freshwater sediment was collected from the Kromme Rijn River at Odijk (The Netherlands). A grabber was used to collect approximately 5 l from the upper 20 cm of sediment. The sediment was stored in a 5 l plastic container and homogenised as much as possible by shaking during 4 d, using an Edmund Bhler (Tbingen, Germany) SM 25 shaker, at a frequency of 200 min−1. The sediment was split into three portions. One portion was used for the characterisation of the sediment, the second was aerated in order to determine the effect of changes in redox conditions on the metal speciation and the third was kept anaerobic.For the aeration, an amount of the homogenised sediment (1 kg) was transferred into a large plastic box (thickness of sediment layer approximately 2 cm) and exposed to the air. The box was constantly shaken at a speed of 200 min−1. The loss of water as a result of evaporation was compensated by adding surface water from the Kromme Rijn River. The aeration continued for 3 weeks, until the concentration of acid volatile sulfide (AVS) had decreased to 0.4% of its original value and was around the detection limit of the applied procedure (0.1 µmol g−1 dry weight). Aeration did not affect the natural pH of the sediment owing to its high buffer capacity (7.3% CaCO3; see Table 2).
Both the aerated and the untreated sediment were used for the sequential extraction procedure. Part of the aerated sediment was acidified with hydrochloric acid to a pH of approximately 5 and the other portion remained at its original pH. For the acidification of the sediment, 0.84 mol l−1 HCl was added continuously to a sediment suspension for a period of 4 h at a speed of 0.1 ml min−1 by means of a Minipuls 2 peristaltic pump (Gilson, Worthington, OH, USA). The suspension consisted of 100 g wet sediment, to which 200 ml of Kromme Rijn surface water were added. The pH of the solution was measured every minute. The solution was stirred continuously during the addition of acid and for 1 h afterwards. The sequential extraction procedure was started after a period of at least 24 h after the acid addition, by which time the pH in the supernatant was constant. The pH level of the natural sediment was 7.46 and 7.52 for the non-aerated sediment and the aerated sediment, respectively. The aerated, acidified sediment had a pH of 5.89.
Sediment characterisation
The sediment was characterised with respect to the following:1. Dry weight content: by drying the sediment for 24 h at 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C.
°C.
2. Carbonate: by volumetric measurement of CO2 gas after addition of acid10 in the dry, ground sediment.
3. Total carbon and sulfur: with a CNS analyser (Fisons Instruments, Milan, Italy, EA 1108) in the dry, ground sediment.
4. “Total" metal contents (Ca, Cd, Cu, Fe, K, Mn, Ni, Pb and Zn): in the dry, ground sediment by means of ICP-AES [Perkin-Elmer (Norwalk, CT, USA) Optima 3000 XL] after digestion with aqua regia. Standard solutions were used for the calibration and internal standards (Ga, In) for quality control. A light sandy soil reference material (BCR, CRM 142R) was used to check the digestion method. The measured metal contents in the reference material were in satisfactory agreement with those certified [Ni, 61.8 mg kg−1 (CRM, 61.1 ± 1.5 mg kg−1); Pb, 25.2 mg kg−1 (CRM, 25.7 ± 1.6 mg kg−1); Zn, 109.6 mg kg−1 (CRM, 93.3 ± 2.7 mg kg−1); Cd, our procedure was not sensitive enough (LOD, 3 mg kg−1) to measure at the level of the reported concentration (CRM, 0.25 ± 0.1 mg kg−1)]. ICP-AES parameters: plasma flow rate, 15 l min−1; nebulizer cross-flow, 0.75 l min−1; auxiliary flow rate, 0.1 l min−1; power, 1400 W; sample uptake, 1.2 ml min−1.
5. AVS: the AVS concentration in the sediment was determined before and after aeration, according to the procedure described by van den Hoop et al.11 The resulting supernatant of the sediment suspension was filtered through a 0.45 µm filter (Millipore, Bedford, MA, USA) and analysed for trace metals by ICP-AES (Perkin-Elmer Optima 3000) and flame AAS (Perkin-Elmer 2100). Standard solutions of the metals in the same matrix as the sample were used for calibration. The sum of the concentrations of Cd, Cu, Ni, Pb and Zn is called simultaneously extracted metals (SEM).
All measurements were performed on duplicate samples.
Sequential extraction
A sequential extraction procedure based on the procedure developed by Tessier et al.7 was used to determine the metal speciation. Six fractions were obtained using various extractants. The fractions, extractants and extraction times are listed in Table 1. After removal of the overlying water of the sediment suspension (which is the water-soluble fraction, F0), 30 g of wet sediment were weighed and used for the first extraction step to obtain F1. The extractions were performed in 250 ml polyethylene centrifuge tubes in order to minimise loss of sediment. The Milli-Q water used for the preparation of the extractants was flushed with nitrogen in order to prevent exposure of the sediment to oxygen. Between each extraction, separation was affected by centrifugation at 3000g for 20 min (Sorvall RC-5B centrifuge, Du Pont Instruments, Wilmington, DE, USA). The supernatant was removed with a 50 ml syringe (Fortuna, Westheim, Germany) and filtered through a 0.45 µm filter (Millipore). The extractant for the next extraction step was added to the remaining sediment in the centrifuge tube. All fractions were analysed for trace metals by ICP-AES (Perkin-Elmer Optima 3000) and flame AAS (Perkin-Elmer 2100). Calibration was carried out using standard solutions. For each sample matrix, a separate set of calibration standards was prepared in the same matrix. The described sequential extraction procedure was performed in duplicate for each of the three samples (untreated, aerated and aerated, acidified sediment). Fraction F5, the residual, was calculated as the difference between the “total" concentration obtained by digestion with aqua regia and the sum of concentrations in F0–F4.| Fraction No. | Extractant | Conditions | Extraction time/h | Fraction description |
|---|---|---|---|---|
| F0 | Overlying water | — | — | Water-soluble |
| F1 | MgCl2 | 60 ml, 1 mol l−1, pH 7 | 1 | Exchangeable |
| F2 | NaAc/HOAc buffer | 60 ml, 1 mol l−1, pH 5 | 5 | Bound to carbonates |
| F3 | NH2OH·HCl | 150 ml, 0.1 mol l−1, pH 2 | 18 | Reducible, bound to Fe/Mn oxides |
| F4 | Oxidation with H2O2 (in HNO3) | 60 ml, 30% (22.5 ml 0.02 mol l−1), pH 2, 85°C |
5 | Oxidizable, bound to organic material and sulfides |
| Extraction with NH4OAc | 37.5 ml, 3.2 mol l−1 | 0.5 | ||
| F5 | “Total" − Σ(F0…F4) | — | — | Residual |
In summary, the procedure in this study was different from the method of Tessier et al.,7 in a few respects. First, wet sediment was used instead of dry sediment, in order to preserve the original metal speciation. An additional extraction step, fraction F0, was introduced which extracts the water-soluble metals. Fraction F5 was calculated from the difference between the “total" metal content determined by aqua regia digestion of the original sediment and the sum of fractions F0–F4, instead of digestion of the sediment remaining after fraction F4 by HF and HClO4. An additional feature of the present study is the aeration and acidification of the sediment in order to study the effect of the change of redox potential or pH on the metal speciation.
Chemicals
All reagents used in this study were of analytical-reagent grade or better. All solutions were prepared using demineralised water, produced by a Milli-Q Reagent Grade system (Millipore). Oxygen was eliminated from the Milli-Q water by purging with nitrogen. The experimental procedure was checked by using a procedure blank. No contamination was detected.Results and discussion
Sediment characterisation
Table 2 gives the results of the sediment characterisation. The relative standard deviation (RSD) of the duplicate measurements is <6%, representing sample reproducibility and analytical reproducibility. The studied freshwater sediment is rich in carbonate and AVS. In Dutch freshwater and marine sediments, AVS contents range from non-detectable (<0.1 µmol g−1) to approximately 50 µmol g−1 dry sediment.11 AVS is present in excess of SEM, which means that the heavy metals could all be bound by sulfides. These results agree with those for a comparable sediment sample from the same site sampled a few years earlier.12 Total concentrations of Ni, Cu, Zn, Cd and Pb are higher than Dutch target values below which no risk to humans and nature is expected. According to Dutch environmental legislation, this sediment can be classified as heavily contaminated, especially because of very high concentrations of Cu, Ni and Zn.| Component | Concentration/µmol g−1a |
“Total" metals (aqua regia digestion) | Concentration/µmol g−1a |
|---|---|---|---|
| a Dry weight. bDifference between total C and carbonate; n = 2. | |||
| Carbonate (as CaCO3) | 7.3% | Al | 1410 |
| Total C | 5917 | K | 179 |
| Organic Cb | 5191 | Ca | 861 |
| Total S | 188 | Mn | 33.7 |
| AVS | 38 | Fe | 914 |
| SEM | 20 | Ni | 1.01 |
| Cu | 1.43 | ||
| Zn | 17.7 | ||
| Cd | 0.05 | ||
| Pb | 1.03 | ||
Sequential extraction
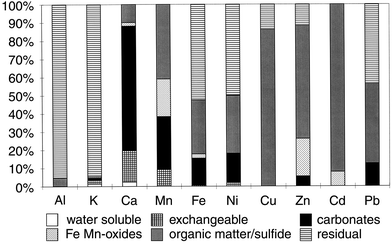 | ||
| Fig. 1 Metal distribution in an untreated freshwater sediment (pH 7.46) as obtained by sequential extraction. Metal contents expressed as a percentage of the total metal content. | ||
 | ||
| Fig. 2 Metal distribution in an aerated freshwater sediment (pH 7.52) as obtained by sequential extraction. Metal contents expressed as a percentage of the total metal content. | ||
Since the residual fraction is not determined experimentally but calculated as the difference between the “total" concentration obtained by digestion with aqua regia and the sum of concentrations in F0–F4, “negative" concentrations may be obtained. “Negative" concentrations in the residual fraction (F5) were calculated for Cd and Ca (15–25 and 2–25%, respectively). For Cd this is probably caused by the inaccuracy of the measurement at concentrations around the detection limit of the applied instrument. Although dry weight determinations showed an RSD of <1% (n = 7), indicating a fairly homogeneous sediment, there might still be differences in Ca content in the different subsamples (e.g., due to shell fragments) which might explain the discrepancy for Ca. If the metal content in the residual fraction is “negative", we assume that the relevant metal does not appear in that fraction and the metal content is considered to be zero. In the calculation of the concentrations in the fractions, a correction was applied for the amount of liquid (extractant + pore water) remaining after the previous extraction.
Aeration of the sediment mainly affects the level of AVS and therefore changes are expected for the metals bound to sulfides: Mn, Fe, Ni, Cu, Zn, Cd and Pb. Al and K predominate in the lattice structure of clay minerals, which will not be dissolved in fractions F0–F4. Al and K are indeed found in the residual fraction. Generally, it is believed that the metals in this fraction will not be available to organisms under normal circumstances. Aeration has no effect on the lattice of clay minerals, which is confirmed by the results of the sequential extraction of the aerated sediment. Ca is mainly present as carbonate. Since protons will be released as a result of oxidation, this causes dissolution of carbonates and Ca becomes slightly more mobile. Aeration hardly affects the distribution of Mn and Ni, although Mn or Ni sulfides are expected to be oxidised and Mn is expected to precipitate as the oxide. This suggests that Ni was originally present in one of the following forms: (a) Ni sulfide and, after oxidation, redistributed and bound to organic matter; (b) bound to organic matter and remaining bound to this phase after oxidation; or (c) bound to both sulfide and organic matter. With this procedure it is not possible to distinguish this. In the literature, it has been suggested that Ni in fraction F4 is most likely bound to organic matter.13 Ni is also partly built into clay minerals, which will not be affected by the aeration.13 Fe becomes less mobile owing to aeration since Fe3+ binds, owing to its higher charge, more strongly to sorption phases such as organic matter than Fe2+. An expected increase in Fe (hydr)oxide precipitates is not observed, possibly owing to the limited time-scale of the experiment. The distribution of Cu and Pb over the fractions seems hardly influenced by oxidation as they are probably mainly bound to organic matter, which is known from literature to occur.13 Zn and Cd, which are for a large part bound to organic matter and sulfides in the untreated sediment, are both released when sulfides are oxidised during aeration of the sediment. After aeration, Zn and Cd are mainly observed in F2, which is related to the sorption to carbonates. Overall it can be concluded that the speciation of Al, K, Mn, Ni, Cu and Pb is hardly affected by aeration. Ca, Zn and Cd are released and subsequently bound as carbonates or adsorbed, whereas Fe is more strongly bound as a result of aeration.
As can be seen from the influence of oxidation on the metal distribution in the sediment, it is important to maintain oxygen-free conditions during extractions of anoxic sediments with respect to fractions F0–F3. In the literature, changes in trace metal contents and a decrease in AVS concentration were observed if no precautions were taken to prevent exposure of anoxic sediment to atmospheric oxygen.6 Sample preservation is also an important factor influencing the partitioning of trace metals in oxic and anoxic sediments. No storage method completely preserves the initial metal partitioning of the sediments. The effects of preservation techniques such as wet storage, freezing, freeze-drying and oven drying on the metal speciation in oxic and anoxic sediments have been studied by several groups. The metal partitioning obtained after pre-treatment was in most cases different from that without pre-treatment of the sediment.4,6,14,15 The changes in speciation observed for metals are associated with the changes in redox chemistry of Fe, Mn, and sulfide. Drying techniques have the most marked effects and should therefore be avoided.6
The results of the sequential extraction are shown graphically in Fig. 3. Acidification of the sediment mainly affects the carbonate content and therefore changes can be expected for the metals bound to carbonates such as Zn, Cd, Ni, Mn and Ca. Since K and Al are lattice elements, no influence of acidification was observed. Carbonates are dissolved when the sediment is acidified, therefore Ca redistributes from the carbonate fraction to the water-soluble fraction. Mn is mobilised during acidification, most likely by dissolution of Mn carbonates. Acidification does not influence the distribution of Fe, Cu and Pb over the fractions as they were only to a small extent bound to carbonates in the aerated sediment at natural pH. In the aerated sediment Zn and Cd were to a large extent present in carbonates which are dissolved during acidification. This causes a shift in the distribution towards more mobile fractions which is also, although to a lesser degree, the case for Ni.
 | ||
| Fig. 3 Metal distribution in an aerated, acidified freshwater sediment (pH 5.89) as obtained by sequential extraction. Metal contents expressed as a percentage of the total metal content. | ||
In conclusion, Ca, Mn, Ni, Zn and Cd, which to a large extent were bound to carbonates in the aerated sediment, become more mobile after acidification. Acidification had hardly any or no effect on Al, K, Fe, Cu and Pb.
| Sediment | Ni | Cu | Zn | Cd | Pb |
|---|---|---|---|---|---|
| Untreated, pH 7.46 | >3.4 | >3.6 | >3.9 | >2.7 | >2.6 |
| Aerated, pH 7.52 | 3.3 | >3.6 | >3.9 | >2.9 | >2.6 |
| Aerated, pH 5.89 | 2.1 | >3.7 | 1.8 | 1.7 | >2.6 |
The applied experimental procedure certainly leads to differences in partition coefficients, as can be clearly seen from the values for aerated acidified sediment and the untreated, anoxic sediment. Kp values in the literature are often given as ranges,16 partly because of differences in applied procedures. For the deduction of quality criteria in the case of absence of toxicity data, it would be necessary to have uniformity in the experimental method for the determination of partition coefficients.
References
- R. M. Smith A. E. Martell, Critical Stability Constants, Vol. 4, Inorganic Complexes, Plenum Press, New York, 1976. Search PubMed.
- G. A. van den Berg, J. P. G. Loch and H. J. Winkels, Water Air Soil Pollut., 1998, 102, 377 CrossRef CAS.
- F. M. G. Tack and M. G. Verloo, Int. J. Environ. Anal. Chem., 1995, 59, 225 Search PubMed.
- M. Kersten U. Förstner, in Chemical Speciation in the Environment, ed. A. M. Ure and C. M. Davidson, Blackie Academic and Professional, Glasgow, 1995, ch. 9, pp. 234–275. Search PubMed.
- P. M. V. Nirel and F. M. M. Morel, Water Res., 1990, 24(8), 1055 CrossRef CAS.
- F. Rapin, A. Tessier, P. G. C. Campbell and R. Carignan, Environ. Sci. Technol, 1986, 20, 836 CAS.
- A. Tessier, P. G. C. Campbell and M. Bisson, Anal. Chem., 1979, 51(7), 844 CrossRef CAS.
- G. Rauret, R. Rubio, J. F. Lopez-Sanchez and E. Casassas, Water Res., 1988, 22, 449 CrossRef CAS.
- J. Kucera, V. Sychra and J. Koubek, Fresenius' J. Anal. Chem., 1998, 360, 402 CrossRef CAS.
- R. H. Loeppert D. L. Suarez, in Methods of Soil Analysis, Part 3, Chemical Methods, ed. D. L. Sparks, SSSA Book Series 5, SSSA, Madison, WI, 1996, pp. 451–455. Search PubMed.
- M. A. G. T. van den Hoop, H. A. den Hollander and H. M. Kerdijk, Chemosphere, 1997, 35, 2307 CrossRef CAS.
- S. E. J. Buykx M. A. G. T. van den Hoop, The Simultaneous Influence of pH and Acid Volatile Sulfide on Metal Speciation in Calcareous Sediments, RIVM Report No. 502501043, RIVM, Bilthoven, 1996. Search PubMed.
- G. Sposito, The Chemistry of Soils, Oxford University Press, New York, 1989. Search PubMed.
- U. Förstner, Int. J. Environ. Anal. Chem., 1993, 51, 5 Search PubMed.
- E. A. Thomsom, Water Air Soil Pollut., 1980, 14, 215 CrossRef CAS.
- N. Drndarski, D. Stojic, M. Zupancic and S. Cupic, J. Radioanal. Nucl. Chem., 1990, 140, 341 CAS.
| This journal is © The Royal Society of Chemistry 2000 |