[60]Fullerene dimers
José L. Segura and Nazario Martín*
Departamento de Química Orgánica, Facultad de Química, Universidad Complutense, E-28040, Madrid, Spain
First published on UnassignedUnassigned11th January 2000
Abstract
The main types of fullerene dimers synthesized so far, including the early all-carbon fullerene dimers, other new carbon allotropes containing two fullerene units and the specially interesting fullerene dimers bridged through electroactive spacers, are reviewed. The different synthetic strategies employed have been divided into two main types, namely: (i) dimerization of C60 itself or some C60 derivatives and (ii) bifunctional cycloaddition reactions to C60.
These new systems are of interest since they can find applications in areas such as artificial photosynthesis, novel molecular electronic devices and in supramolecular chemistry.
 José L. Segura José L. Segura | Nazario Martín studied chemistry at the Universidad Complutense de Madrid (UCM), where he obtained his PhD under Professor C. Seoane in 1984. After a year working on X-ray contrast agents in a pharmaceutical company, he worked as a postdoctoral fellow (1987–1988) at the Institut für Organische Chemie der Universität Tübingen under Professor M. Hanack on electrically conducting organic materials. In 1994, he was a visiting Professor at the Institute for Polymers and Organic Solids (IPOS) at the University of California, Santa Barabara (UCSB) working with Professor F. Wudl on modified fullerenes. Since 1989, he has been Associate Professor of Organic Chemistry at the University Complutense (UCM). His research interests range over the chemistry of fullerenes and electroactive molecules with emphasis on the electrical and optical properties. He is also active in the asymmetric synthesis of heterocyclic systems. |
 Nazario Martín Nazario Martín | José L. Segura received his education in chemistry at the Universidad Complutense de Madrid, where he obtained his diploma in 1989 and his PhD in 1994 under Professor N. Martín and Professor C. Seoane. In 1993 he visited the group of Professor W. Daily (Univesity of Pennsylvania) where he worked in the area of strained-ring organic syntheses. After post-doctoral stays in the group of Professor M. Hanack (University of Tübingen) working in the synthesis of electroluminescent conjugated polymers and in the group of Professor F. Wudl (University of California at Santa Barbara) working on the functionalization of C60 fullerene, Dr Segura joined the faculty in the Department of Organic Chemistry at the Universidad Complutense de Madrid. |
1 Introduction
Connecting fullerene cages has been a subject of interest since the observations of fullerene coalescence upon laser irradiation of C60 films.1 The experimental evidence for molecular coalescence of C60 molecules, that is, formation of stable higher fullerenes which are multiples of the initial masses, is clearly manifested by measurements of the mass spectra where C120 is the most abundant product. However, the shapes of the products from the coalescence reactions are not clearly known and it is not clear whether they are formed as large clusters or as dumbell-shaped (C60)2 dimers.1 While the coalescence products are too complex to be analyzed, the seemingly simple addition polymers have not been well characterized either. Thus, as dimeric fullerenes have been proposed as essential subunits of the all-carbon fullerene polymers, it has been of great importance to clarify their detailed structure as well as their physical and chemical properties for a better understanding of the nature of fullerene polymers.2 Whereas early efforts were mainly directed at the obtention of all-carbon fullerene dimers in the search for new carbon allotropes, with the development of fullerene chemistry it has been possible to obtain properly functionalized carbon spheres in order to synthesize a variety of the so called dumbbell type dimeric materials with different spacers between the fullerene units. In this article we present an overview of the main types of [60]fullerene dimers synthesized so far, putting particular emphasis on the different synthetic strategies followed for their obtention. These strategies will be divided into two main classes: (i) dimerization of either fullerene itself or a suitably functionalized fullerene and (ii) bifunctional cycloadditions to [60]fullerene.On the other hand, the design of novel organofullerenes containing electroactive moieties constitutes a promising field due to the interesting optical and electronic properties they can display. Another approach to this field has been the study of fullerene dimers in which two fullerene spheres are attached either directly or are covalently connected through a molecular electroactive bridge. The electronic interactions between the C60 units themselves as well as between the C60 units and the electroactive spacer have been the subject of different studies and we will look especially at the electrochemical and photophysical properties of these redox active compounds.
2 Dimerization reactions of [60]fullerene and derivatives
C120 is the most abundant product present in the mass spectra of coalescence experiments as well as in different polymerizations. Therefore, theoretical and experimental studies have been carried out in order to determine its ground-state configuration. These studies reveal that, in the neutral state, the energetically most favourable bonding between the two C60 molecules is accomplished through a [2 + 2] cycloaddition, where the (6,6) short bonds between two hexagons on adjacent molecules are broken and converted into a four membered ring (1, Fig. 1)3 with D2h symmetry.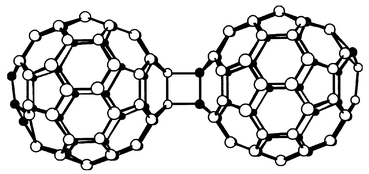 | ||
| Fig. 1 | ||
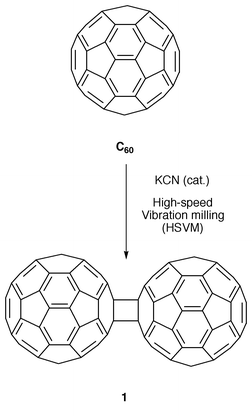
Therefore, 1 has been proposed as the smallest subunit of the all-carbon fullerene polymers produced by coalescence reactions on [60]fullerene. In contrast to the many theoretical studies carried out on this compound, only recently have efficient methods for its synthesis been reported.2 Thus, the bulk synthesis of 1 was achieved by the solid state mechanochemical reaction of C60 with KCN by using a high speed vibration milling (HSVM) technique (Scheme 1). Two different mechanisms have been proposed for this reaction: (i) dimerization via nucleophilic addition and (ii) dimerization via coupling through an electron-transfer step.3
 | ||
| Scheme 1 | ||
Dimer 1 is a dark brown powder which is barely soluble in most organic solvents. The solubility depends greatly on the purity: the purer the sample, the less soluble it is. It was found to dissociate quantitatively into C60 when heated at 175 °C for 15 min and also to undergo slow photochemical dissociation into C60 under exposure to room light. 0.8% of C60 is produced when a powdery sample of 1 is subjected to a hydraulic pressure of 100 kg cm−2 for 30 min. The cyclic voltammogram of 1 exhibits three reversible reduction waves which are almost identical to those of C60 observed under the same conditions. A slight shoulder on the first reduction peak of 1 is the only remarkable difference to C60, thus indicating that dimer 1 readily dissociates into two C60 units immediately after C120 acquires extra negative charge.3 The structure of 1 has been determined by X-ray crystallography for a single crystal grown in an o-dichlorobenzene (ODCB) solution.2 The dimer is composed of two C60 cages sharing a cyclobutane ring with the C120 molecules arrayed in highly ordered layers, different from the face-centered cubic arrangement of C60. The relative elongation of the inter-cage bonds (1.575(7) Å) compared with the ordinary C(sp3)–C(sp3) single bond may be responsible for the above mentioned ready thermal cleavage of 1.
The close similarity between the FT-IR spectrum of all-carbon poly[60]fullerenes and 1 has been used as evidence for the proposed [2 + 2] cycloaddition linking pattern in polyfullerenes.
Other extensively studied fullerene dimers are fullerene oxides such as C120O. The synthesis of this type of compounds was stimulated by the discovery of the odd-numbered fullerene clusters4 and Taylor’s proposed mechanism for the generation of C119via thermal decarbonylation of C60O and addition of the resultant C59 to C60.5
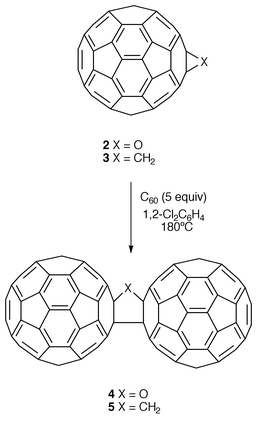
In 1995 it was almost simultaneously reported by two independent groups6,7 that thermolysis of C60O with a [6,6] epoxide structure (2) in the presence of C60 affords the corresponding oxygen-bridged C60 dimer (4). By using the same experimental conditions, the methylene-bridged C60 dimer (5) was also obtained by thermolysis of the [6,6] fullerene cyclopropane 3 and C60 (Scheme 2).
 | ||
| Scheme 2 | ||
Very recently, Taylor and co-workers have shown that at ambient temperature and in the solid state C60 degrades to C120O (4) which is present in up to 1% concentration in the samples examined and they have been able to extract multimilligram quantities of C120O (which crystallizes from carbon disulfide as black needles) from commercial C60.8 The authors claim that C60 undergoes oxidation to C60O (2) on exposure to air, but given that the mono-oxide is strained and is predicted to be unstable relative to C120O, it is captured by a further C60 molecule in a [2 + 2] reaction to produce C120O (4).
In the electrochemical study of dimer 4 formed by two C60 cages connected by a tetrahydrofuran ring, two reversible asymmetric peaks along with a resolved set of two reversible peaks can be observed. Each asymmetric peak corresponds to two closely spaced waves resulting from the superposition of two processes with very close reduction potentials. This electrochemical behaviour indicates that the two cages are not reduced simultaneously, thus suggesting that the two C60 moieties are communicating, in contrast to what is observed for dimer 1 where the two C60 units are connected by a cyclobutane ring.
Following thermal reactions of the C60/C60O/C60O2 system in the solid state, various other dimeric fullerene derivatives such as C120O2 (6) and C119 (7) have been isolated and characterized. In C120O2 the links are composed of two furanoid rings sharing a C–C bond interconnecting with a central 4-membered ring.9 Here, in contrast to C120O, the four-membered ring is thought to connect between single (6,5) bonds of the original C60’s (Fig. 2).
 | ||
| Fig. 2 | ||
More recently, the thermolysis of C120O has been performed in the presence of sulfur yielding the corresponding sulfur analogue C120OS (8).10 This is the first sulfur containing dimeric [60]fullerene derivative to be synthesized and characterized by spectroscopic methods. The data obtained are consistent with the lowest-energy modeled structure in which two fullerene cages are linked via furan and thiophene bridging rings with a cis configuration of the 6,6 junctions (Fig. 3).10
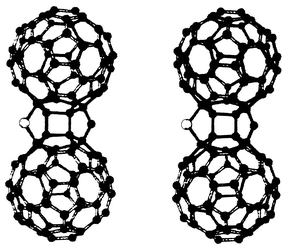 | ||
| Fig. 3 | ||
After early mass spectroscopic work proved that odd-numbered dimer-related species can be formed out of fullerene epoxides,5 it has proven possible to isolate C119 (7). The ‘classical’ fullerenes contain only an even number of quasi-sp2 carbon atoms in a closed-cage structure in which pentagons are completely surrounded by hexagons. However, McElvany et al. discovered the presence of odd-number clusters C119, C129 and C1394 in the thermal desorption mass spectra of a toluene extract of comercial soot and in the spectra of the products of ozone–fullerene reactions. These species can be obtained by thermal decomposition of C120Ox (x < 3). C119 is oxygen sensitive and decomposes within hours under ambient conditions although under argon is stable in a refrigerator for weeks. C119 is thought to consist of two partially opened cages connected by three sp3 carbons and two seven-membered rings (Fig. 4). Consequently, although it retains a high degree of topological analogy to the title dimers, the structural integrity of the individual cages is no longer maintained in C119.
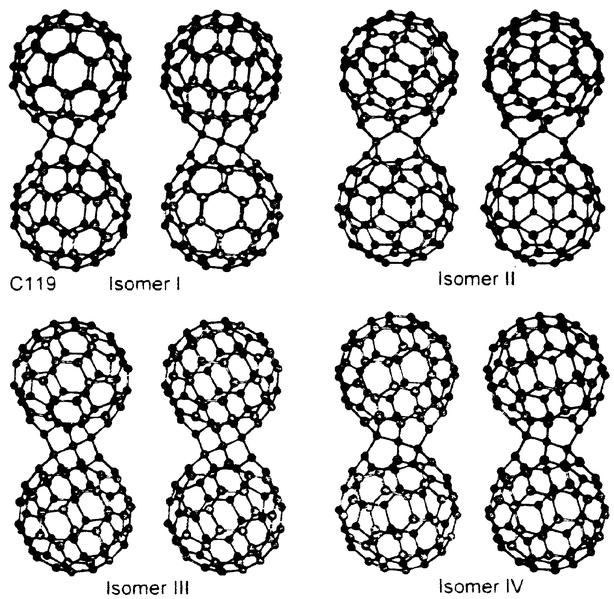 | ||
| Fig. 4 | ||
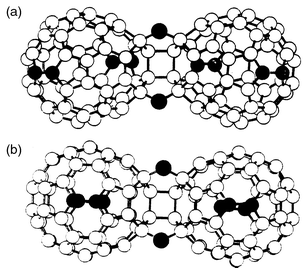
As was previously stated, in the neutral case, the energetically most favourable bonding between the two C60 molecules is accomplished through a [2 + 2] cycloaddition. Thus, the energy calculated for a single bonded (SB) isomer (9) formed by direct covalent bonding between two C60 molecules is higher than that obtained for 1. With the addition of one electron to each C60 molecule, however, different calculations11 indicate that the relative energy ordering between the [2 + 2] and SB structure reverts. As a result, the SB dimer is predicted to be more stable than the [2 + 2] isomer in mono-anionic fullerides. Recently, X-ray results show that quenching the high-temperature face-centered cubic (fcc) phase of A1C60 (A = K, Rb) below 280 K produces a metastable orthorhombic structure in which the molecules exist as dimer pairs. The experimentally observed 9.34 Å center-of-mass distance in the charged dimer is in good agreement with the calculated value of 9.30 Å for the SB configuration with a C2h symmetry (the two balls being in a trans position, Fig. 5a).11 Interestingly, these dimers resulting from the monodoped fulleride structures (C1202−) are isoelectronic with the azafullerene dimer (C59N)2 (10) which has been shown to be bonded by a single C–C intermolecular bond (Fig. 5b).12
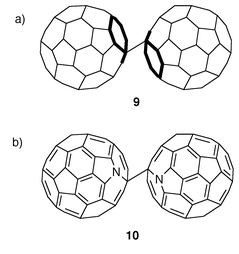 | ||
| Fig. 5 | ||
Some of the best studied fullerene dumbbell-shaped dimers are those with the general structure RC60C60R (11 R = H, halo, alkyl, fluoroalkyl) which are formed by the recombination of RC60· radicals, which in turn can be prepared in situ by addition of R· to C60.13,14 (Fig. 6).
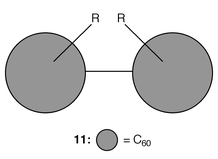 | ||
| Fig. 6 | ||
An important aspect of the chemistry of C60 is its reactivity towards free radicals. Radicals add rapidly and multiply to the double bonds of C60 to give products RnC60. A remarkable feature of the EPR spectra of the RC60 adducts is the dramatic increase in intensity of the spectrum with increasing temperature, in contrast to the usual decrease in intensity associated with the Curie law. Temperature dependence of this type may be indicative of an equilibrium between a radical and its dimer. Thus, the dependence of signal intensity on temperature and dilution at a fixed temperature established that the radical adducts of C60 exist in equilibrium with their dimers and that the dimer bond strength depends on the size of the group R.13
| RC60C60R ⇌ 2RC60 | (1) |
Substituents R are considered to be attached at ortho or para positions relative to the intercage bond, based on the EPR spectra of monomeric species RC60· showing the unpaired electron localized within the six membered ring bearing the R group.13 C–C single bonds bearing large cage fragments at both ends provide interesting targets for studies of internal rotation and rotational isomerism. Thus, Osawa and coworkers have studied the rotation about the intercage bond of singly bonded RC60C60R (R = H, Me, and t-Bu attached at the ortho or para positions from pivot carbon atoms).14 They observed that rotamers with R groups situated close to each other as in the gauche form are predicted to be more stable than those with maximally separated R groups (trans) and rationalized the unexpected stability of the gauche-like conformation in terms of a lever effect peculiar to the dumbell shape of the molecule.14
An interesting example of an RC60C60R dimer, in which R is a morpholine group (11, R = morpholine) has been reported by Hirsch and co-workers.15 This first aminated fullerene dimer with a single inter-fullerene connection was isolated, together with dehydrogenated amino adducts with a preferred 1,4-addition pattern, in the reaction of [60]fullerene with an excess of morpholine in benzene in the presence of oxygen. The authors claimed that the fact that only dehydrogenated products were isolated shows that the initially formed intermediates such as aminated fullerene carbanions R2NC60− or zwitterions R2HN+C60− are oxidized by oxygen to form aminated radicals R2NC60· which may recombine through a dimerization process (Fig. 6).
A different approach towards the obtention of a new kind of carbon allotrope involves the combination of fullerene and acetylene units in the so called all carbon chemistry. Among the attractive intermediates on the route towards these new carbon allotropes are the cyclic polyynes (12, 13) depicted in Fig. 7 in which macrocyclic polyynes are ‘end-capped’ with methanofullerenes.16
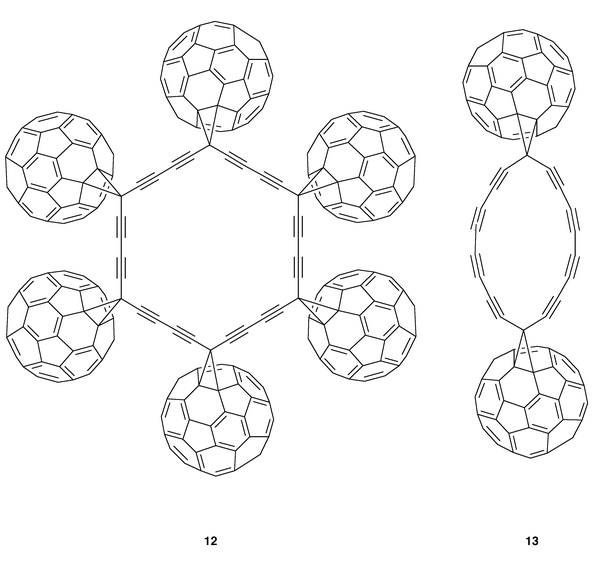 | ||
| Fig. 7 | ||
With this aim, Diederich and co-workers developed new synthetic routes to symmetrically and unsymmetrically substituted diethynylmethanofullerenes which have been used in the preparation of dimeric fullerene derivatives. Thus, monoprotected diethynylmethanofullerene (14) was homocoupled under Hay coupling conditions to the corresponding butadiynyl-linked dimeric methanofullerene (15, Scheme 3) which was isolated in 37% yield as black shiny crystals, reasonably soluble in cyclohexane, toluene and CS2.
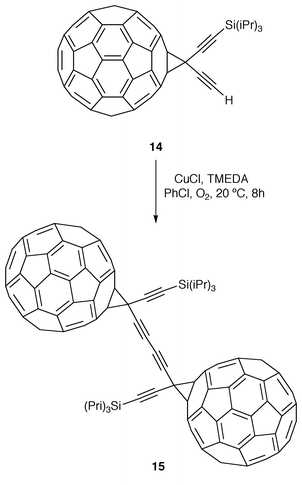 | ||
| Scheme 3 | ||
In the cyclic voltammetry measurements performed on 15, a single reduction potential value (−0.99 V, in CH2Cl2vs. Fc/Fc+) assigned to the transfer of one electron to each of the two C60 moieties and two very similar potentials (−1.43 and −1.56 V) for their second reduction steps were observed. These values are similar to those observed for the monomeric analogs, thus suggesting that both through-bond and through-space electronic interactions between the two fullerene moieties in 15 are not very efficient. This fact is in agreement with the UV/Vis data16 as well as with the X-ray structural data17 and indicates that the C(sp3) atoms in the two butadiynediyl-linked methano bridges of 15 act as insulators.
Alternatively, dimeric fullerene derivatives in which the acetylene spacers are directly attached to the fullerene core have been prepared. Diederich and co-workers have recently synthesized buta-1,3-diynediyl-linked bis(fullerenes) (17, 18) and have made attempts at their conversion into the corresponding dianion 19.18 Syntheses of these compounds were carried out by homocoupling of ethynylfullerenes 16, bearing base-stable substituents at their 2-position. In situ preparation of the Hay catalyst [CuCl–TMEDA–O2] (TMEDA = N,N,N′,N′-tetramethylenedi amine) in PhCl in the presence of 16 followed by stirring for 24 h in dry air at room temperature afforded bisfullerenes 17. Removal of the tetrahydropyran-2-yl (THP) protecting groups in 17b gives the highly insoluble dumbbell 18 with hydroxymethylene groups on the 2,2′-positions of the buta-1,3-diynediyl-bridged carbon spheres. Attempted conversion of 18 to the all-carbon dianion 19 (C1242−) via base-induced elimination was not successful18 (Scheme 4).
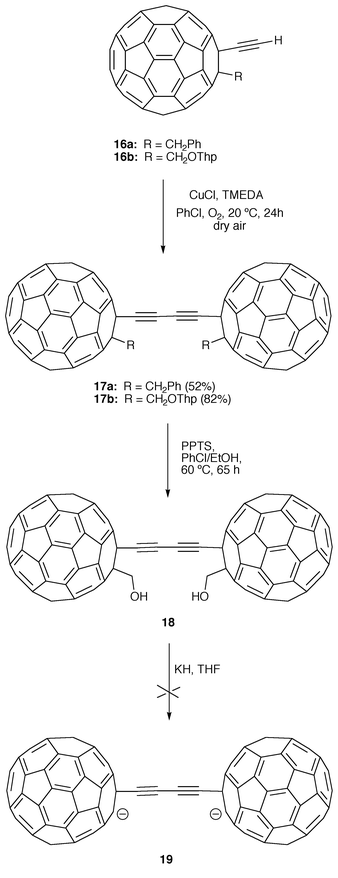 | ||
| Scheme 4 | ||
As observed for the parent dumbbell fullerenes 15, the cyclic voltammetry measurements performed for 17 clearly indicate that the two C60 moieties behave as two electrochemically independent units.18
Komatsu and co-workers have synthesized, in a very low yield, a fullerene dimer with an acetylene spacer (20), instead of the butadiyne linker present in 17, by reaction of dilithioacetylene with C60 in toluene, followed by addition of dodecyl iodide at reflux temperature (Scheme 5).19
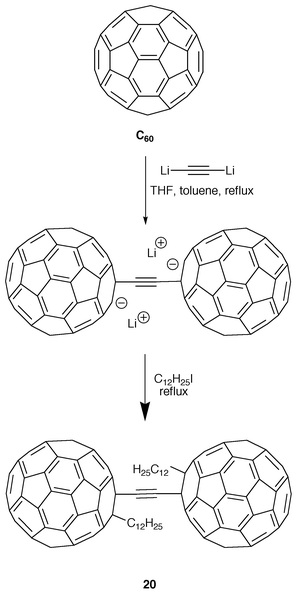 | ||
| Scheme 5 | ||
As in the preceeding examples, there is no evidence for electronic interaction between both C60 cages observable in the electronic spectra of 20. Cyclic voltammetry measurements performed on 20 show that fullerene moieties are simultaneously reduced, thus confirming the absence of electronic interaction between them.19
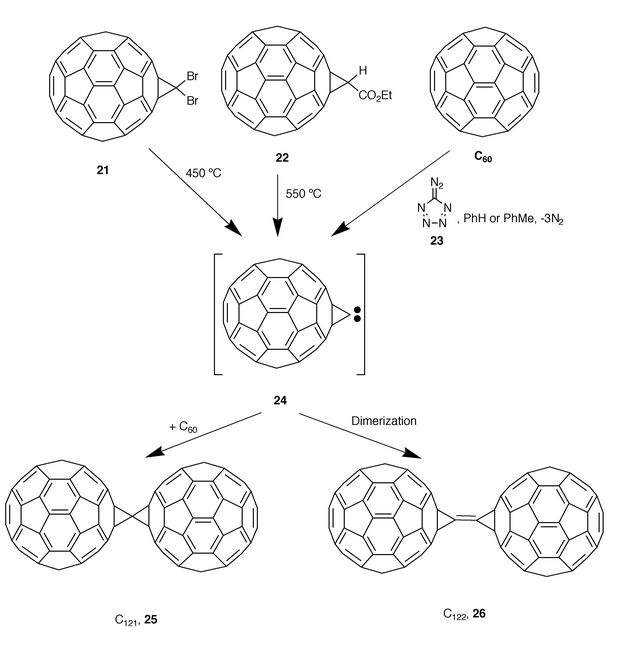
A recent and outstanding study on the dimerization of methanofullerenecarbene, C60C: (24) and its addition to C60 has led to the new carbon allotropes C121 (25) and C122 (26) (Scheme 6).20
 | ||
| Scheme 6 | ||
Carbene intermediate 24, produced from the methanofullerene 21, was proposed as a precursor of C121 and C122 during pioneering studies by Osterodt and Vögtle, which were detected by mass spectrometry.21 In 1998, Strongin and co-workers reported the first isolation and characterization of C122via the reaction of [60]fullerene with the atomic carbon precursor diazotetrazole 23.22 It was isolated as a black solid that formed a golden-yellow solution in toluene. It showed two intense characteristic fullerene absorptions at 210 and 260 nm, while no clear absorptions in the region from 400 to 700 nm were observed. Spectroscopic studies are consistent with a dimeric D2h-symmetric structure. Dragoe and co-workers have also succeeded very recently in the syntheses of both C121 and C122.20 When a mixture of C60CBr2 (21) and C60 was heated at 450 °C in an argon atmosphere at a rate of 5 K min−1 and then cooled to room temperature, a black powder, soluble in CS2 and o-dichlorobenzene was obtained (the same product could be obtained by using C60CHCO2Et 22 instead of C60CBr221 and a higher temperature, i.e. 550 °C). The mixture was chromatographed (GPC, toluene) and both C121 and C122 could be isolated. The amount of C122 decreases when increasing the C60/C60CBr2 ratio; the formation of C122 may be suppressed when using more than a ten-fold excess of C60. Spectroscopic data obtained for C121 are in agreement with a C2v symmetry for the molecule. Scanning tunneling microscopy (STM) measurements on samples of both C121 and C122 show similar images corresponding to dumbbell-like structures.20
3 Bifunctional cycloadditions to [60]fullerene
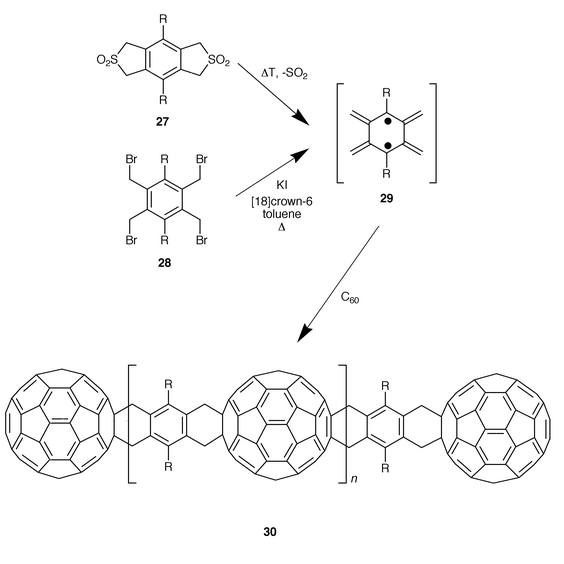
A large variety of cycloadditions have been carried out with C60 and the complete characterization of both monoadducts and multiple adducts greatly increased the knowledge of fullerene chemistry. [60]Fullerene is an excellent dienophile in cycloaddition reactions which reacts by a [6,6] double bond, thus avoiding the unfavourable [5,6] double bonds within the fullerene skeleton. Cycloadditions to C60 have been well-studied, running from [2 + 1], [2 + 2], [3 + 2], [4 + 2] to [8 + 2], yielding the respective monocycloadducts which are easily isolated in moderate to good yields. In contrast, bisaddition leads to the formation of regioisomeric mixtures of bis-adducts which are usually very difficult to separate. Derivatization of C60 with bifunctionalized molecules has been used as an alternative and efficient method for the preparation of fullerene dimeric materials.Wudl and co-workers reported in 1992 the first syntheses of dimers by reaction of bis(diazoderivatives).23 Since then, different dumbbell-like fullerene dimers have been synthesized by using bisdienes in Diels–Alder cycloadditions. In 1993, Müllen and co-workers reported that bis-o-quinodimethane 29 (R = H), generated in situ from 1,2,4,5-tetrakis(bromomethyl)benzene (28 R = H), indeed captures two fullerene molecules to provide a highly insoluble bisadduct (30, n = 0, R = H, Scheme 7).24 When flexible, solubilizing substituents were introduced in the bis-o-quinodimethane moiety (29 R = OHex) different oligomeric materials 30 (n = 0–5) could be detected by using MALDI-TOF mass spectrometry.25 The dumbbell compound 30 (n = 0, R = OHex) was found to be soluble in halogenated benzenes and exists as a single regioisomer so that it was possible to characterize it by NMR spectroscopy.
 | ||
| Scheme 7 | ||
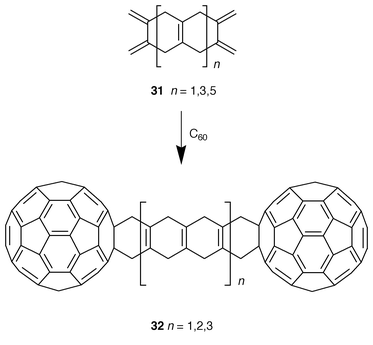
Paquette and co-workers have also used bisdienes to prepare dumbbell-like systems 32 in which a pair of C60 units are linked through cyclohexa-1,4-dienyl ladders of different length (Scheme 8).26 With control of the spacing of the spheres, it is expected to gain some knowledge of the possible operation of intramolecular communication between the fullerene cages. However, a restriction of exploitation of these systems is their striking insolubility.
 | ||
| Scheme 8 | ||
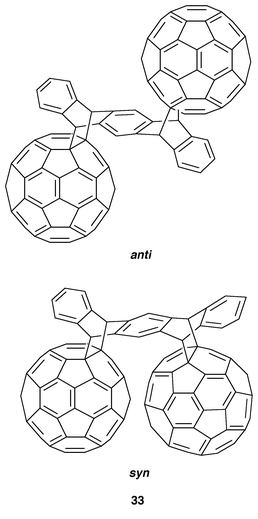
More recently, a solid state [4 + 2] cycloaddition of pentacene to C60 using a high-speed vibration milling (HSVM) technique afforded a novel C60 dimer (33) with the two C60 cages assumed to be in the less sterically hindered anti-face arrangement.27
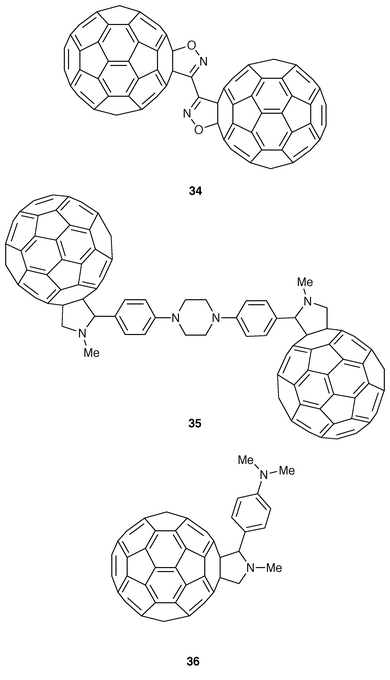
Other selected fullerene dimers obtained from bifunctional cycloadditions are given in Fig. 9. Dimer 34 was obtained from the double 1,3-dipolar cycloaddition of [60]fullerene to cyanogen di-N-oxide (Fig. 9).28 The solubility of 34 in a mixture of 1-chloronaphthalene–2-chloronaphthalene 9∶1 (v/v) allowed its characterization by recording 13C-NMR spectra. The cycloaddition occurs at the [6,6] ring junction of the fullerene and the number of signals clearly indicate a Cs symmetry of the molecule.
 | ||
| Fig. 8 | ||
 | ||
| Fig. 9 | ||
1,3-Dipolar cycloadditions of azomethine ylides to C60 is a suitable and quite general procedure for the functionalization of [60]fullerene.29 In 1996, we reported the first synthesis of the dimeric [60]fullerene 35 (Fig. 9) by using this type of cycloaddition reaction.30 Thus, by reacting C60 with sarcosine and the appropriate dialdehyde in refluxing toluene, 35 was isolated as a black solid showing an extremely low solubility in most organic solvents. Its UV-Vis spectrum shows the presence of a band at around 410 nm, in addition to the typical weak absorption band at 430 nm of dihydrofullerenes, thus confirming the [6,6]-closed character of the dimer. The cyclic voltammetry measurements indicate that there is no significant interaction between both fullerene cages of the dimer in the ground state, as has been previously observed for dyad 36.30
4 Fullerene dimers connected through molecular electroactive bridges
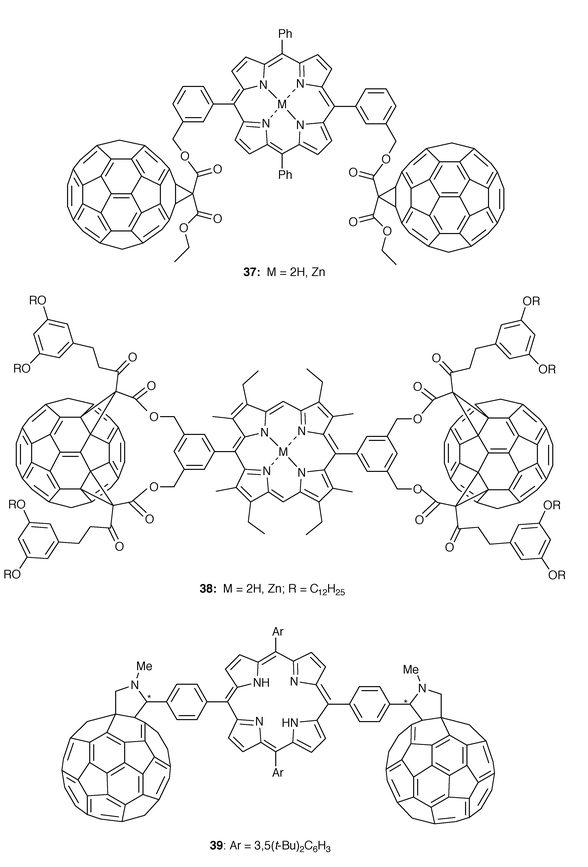
As stated in the Introduction, the design of dyads bearing covalently linked electron donor moieties in close proximity to the C60 surface is a valuable approach to artificial photosynthesis and novel molecular electronics. We have recently reviewed the wide variety of C60 donor dyads in which the C60 core is covalently attached to an electron donor unit as promising candidates for the search of electron-transfer properties.31 An intriguing and less studied approach consists of the synthesis of fullerene dimers in which the two fullerene spheres are connected through a molecular electroactive bridge. The electronic interactions between both C60 units as well as the C60 units and the electroactive spacer have been the subjects of different studies.Intramolecular processes such as electron and energy transfer have been observed in different C60–porphyrin dyads. Very recently, triads C60–porphyrin–C6037, 38, and 39 (Fig. 10) have been synthesized, and their electronic properties studied.
 | ||
| Fig. 10 | ||
Triads 37 and 38 have been synthesized by using Bingel macrocyclization of porphyrin tethered bismalonates32 while 39 has been obtained by 1,3-dipolar cycloaddition from the corresponding formylporphyrins, N-methylglycine and C60 in toluene.3338 is obtained as a mixture of two conformers in slow equilibrium, as shown by a variable temperature NMR study and represents the first example of a bis(meta-substituted phenyl)porphyrin for which the barrier to free rotation is high enough at room temperature to be able to distinguish the cis and trans isomers. On the other hand, because of the unsymmetrical carbon of the pyrrolidine units, two stereoisomers corresponding to the meso and racemic isomers could be isolated for 39. A strong quenching of the flurescence emission of porphyrin is observed for the three triads (37-39). From the electrochemical measurements, it can be stated that the mutual electronic effects exerted by the fullerene on the porphyrin and vice versa in 39 are relatively small and, thus, the ground-state donor–acceptor interactions are hardly measurable by electrochemical procedures.32
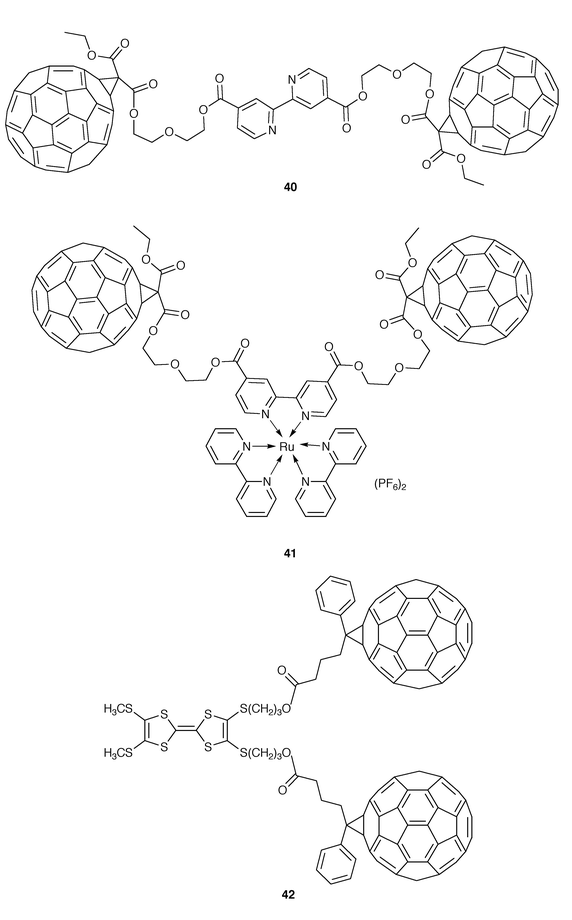
Other electroactive bridges used to connect fullerene moieties are bipyridine (bpy) ligands and the strong electron donor tetrathiafulvalene (Fig. 11). Very recently, the synthesis of the bis-C60–bpy derivative 40 has been reported.34 Functionalization of C60 by using the Bingel reaction was followed by a multistep synthetic pathway to yield the fullerene dimer 40 as a soluble material in which the methanofullerene moieties are separated from the bipyridine ligand by polyethoxy spacers. The subsequent reaction of 40 with [Ru(bpy)2Cl2] yields the ruthenium(II) complex [Ru(40)bpy2][PF6]2 (41). The electronic absorption spectrum of this triad displays absorptions assigned to the ruthenium complex subunit and the fullerene moieties. The observed data suggest the absence of charge transfer between both units.
 | ||
| Fig. 11 | ||
Fullerenes covalently attached to the strong electron donor tetrathiafulvalene (TTF) moieties are very attractive systems for the preparation of photovoltaic devices. The TTF moiety forms, upon oxidation, thermodynamically very stable radical-cation and dication species due to the aromatic character of the 1,3-dithiolium cations. This gain in aromaticity represents a new concept in stabilization of the charge-separated state in electroactive dyads. Although different attempts have been made to synthesize C60–TTF–C60 triads, to the best of our knowlege, only one example is reported in the literature.35 Delhaès and co-workers have prepared three molecular assemblies in which a TTF derivative is covalently linked to two (42, Fig. 11) or even four C60 molecules through flexible rings. UV-Vis spectroscopy as well as cyclic voltammetry measurements indicate that no interaction between the different donor and acceptor systems of the supermolecules can be detected in the ground state. Nevertheless, these systems are promising candidates for further photophysical studies in the search for improved optoelectronic devices.
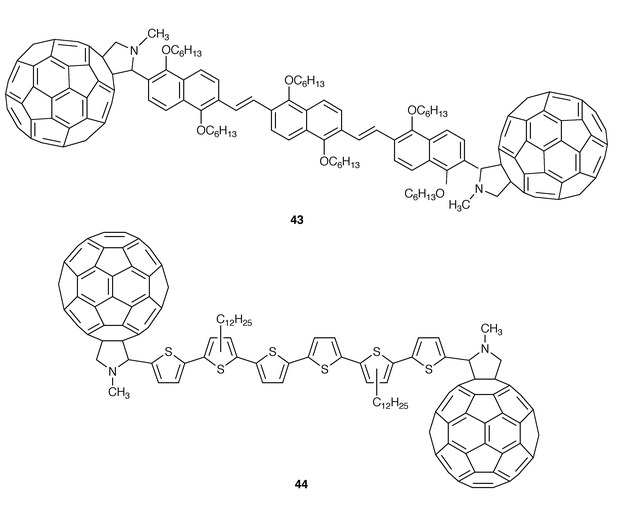
The photoinduced electron transfer from conducting polymers derived from polyphenylenevinylenes as well as polythiophenes to C60 has been used to prepare improved polymeric photovoltaic cells with high efficiency. These polymer–C60 systems represent an outstanding and realistic application of fullerenes. While the forward electron transfer reaction occurs in a subpicosecond time scale, the recombination of the photogenerated electrons on fullerenes and the holes in the polymer is slow (μs). The study of photoinduced electron transfer in molecular dyads and triads of conjugated oligomers covalently linked to fullerenes has been proposed in order to investigate whether this large difference in electron transfer rates is an intrisic property of the structures involved. To the best of our knowledge, only two examples of conjugated oligomers end-capped with two C60 moieties (43, 44, Fig. 12) have been reported and both have been prepared by 1,3-dipolar cycloaddition from the corresponding diformyl oligomers, N-methylglycineand C60.36,37 We have synthesized molecular triads of oligo-2,6-naphthylenevinylenes end-capped with [60]fullerene 4336 and observed that the strong fluorescence of the conjugated oligomer is strongly quenched by the fullerene moiety in the triad. Similarly to the porphyrin analogues, the electrochemical study of 43 reveals that no interaction takes place between the conjugated oligomer and the fullerene moiety in the ground state. It is important to note that oligomers are well-defined molecules which enable a relationship to be established between the structural effects and electronic properties of the related polymers and, therefore, the C60-oligomer-C60 systems deserve to be studied in more depth.
 | ||
| Fig. 12 | ||
Finally, with the aim of incorporating fullerenes into multicomponent molecular systems in order to provide access to the construction of two-and three-dimensional supramolecular architectures with defined geometries, some dimeric fullerenes have been prepared. Thus, taking advantage of the Pt(II)-directed self-assembly of multidentate ligands, a tetracationic cyclophane 45 containing two fullerene moieties has been recently synthesized as a tetrakis-triflate salt (Fig. 13).38 The X-ray analysis of this structure indicates the almost quantitative nature of the self-assembly process. The use of this strategy paves the way for building up complex supramolecular fullerene arrays.
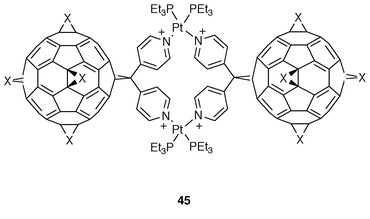 | ||
| Fig. 13 | ||
On the other hand, a copper(I)-complexed rotaxane (46, Fig. 14) bearing two fullerene stoppers has been prepared in an interesting extension of the use of electro- and/or photoactive species in the preparation of multicomponent molecular systems displaying electronic and photochemical properties.39 Rotaxane 46 was prepared by using the above studied procedure of oxidative coupling reaction of terminal alkynes.16 Electrochemical studies carried out on 46 indicate a strong effect of the fullerene moiety leading to a significant anodic shift of the CuI/II redox potential (+0.865 V vs. SCE in CH2Cl2) as compared to other related complexes (ΔE° ≈ 0.2 V). In contrast, the metal center does not seem to change the redox properties of the fullerene units which show the first reduction potential at around −0.6 V. Luminescence measurements show that emission from the metal-to-ligand charge-transfer triplet state of the copper(I) complex 46 is quenched by the two C60 units as stoppers.
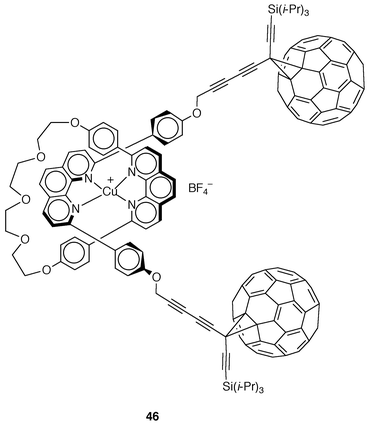 | ||
| Fig. 14 | ||
5 Summary and conclusions
In this article, we have presented selected new classes of dimeric fullerenes in which the carbon spheres are connected in a variety of ways.Early efforts in this area were mainly directed to the obtention of all-carbon fullerene dimers and polymers in the search for new carbon allotropes by using physical methods such as laser irradiation or high pressure transformations. However, the products obtained were too complex to be analyzed and many of the isolated products could not be properly characterized.
With the development of covalent fullerene chemistry, new molecular carbon allotropes containing two fullerene cages such as C119, C120, C121 and C122 could be prepared by means of different synthetic procedures. Spectroscopic and crystallographic studies have allowed the identification of their structures which has been of great importance in understanding the nature of all-carbon fullerene polymers.
The major advantage of the synthetic pathways toward dimeric fullerenes is the possibility of introducing functional groups which allows for the obtention of new types of soluble dumbbell fullerene dimers with very different structures and properties. The different synthetic strategies employed have been divided into two main types, namely: (i) dimerization of C60 itself or some C60 derivatives and (ii) bifunctional cycloaddition reactions to C60.
Depending upon the spacer between the fullerene cages, different degrees of interaction among them, either in the ground or in the excited states, have been observed by using different electrochemical and photophysical methods. Specially interesting are fullerene dimers connected via molecular electroactive bridges as they can find applications in areas such as artificial photosynthesis and novel molecular electronic devices.
Finally, with the aim of incorporating fullerenes into multicomponent molecular systems in order to provide access to the construction of two- and three-dimensional supramolecular architectures with defined geometries, some dimeric fullerenes have been prepared.
In conclusion, C60 dimers represent a new class of carbon allotropes and new modified fullerenes whose spheres can be either directly linked or connected through a functionalized spacer. Both supermolecules are promising candidates for the still demanding application of fullerenes.
Acknowledgements
We thank the DGICYT of Spain (Project PB95-0428-CO2) for financial support.References
- C. Yeretzian, K. Hauser, F. Diederich and R. Whetten, Nature, 1992, 359, 44 CrossRef CAS.
- G.-W. Wang, K. Komatsu, Y. Murata and M. Shiro, Nature, 1997, 387, 583 CrossRef CAS.
- K. Komatsu, G.-W. Wang, Y. Murata, T. Tanaka, K. Fujiwara, K. Yamamoto and M. Saunders, J. Org. Chem., 1998, 63, 9358 CrossRef CAS.
- S. W. McElvany, J. H. Callahan, M. M. Ross, L. D. Lamb and D. R. Huffman, Science, 1993, 260, 1632 CAS.
- R. Taylor, J. Chem. Soc., Chem. Commun., 1994, 1629 RSC.
- A. B. Smith III, H. Tokuyama, R. M. Strongin, G. T. Furst and W. J. Romanow, J. Am. Chem. Soc., 1995, 117, 9359 CrossRef.
- S. Lebedkin, S. Ballenweg, J. Gross, R. Taylor and W. Krätschmer, Tetrahedron Lett., 1995, 36, 4971 CrossRef CAS.
- R. Taylor, M. P. Barrow and T. Drewello, Chem. Commun., 1998, 2497 RSC.
- A. Gromov, S. Lebedkin, S. Ballenveg, A. G. Avent, R. Taylor and W. Krätschmer, Chem. Commun., 1997, 209 RSC.
- S. Giesa, J. H. Gross, W. E. Hull, S. Lebedkin, A. Gromov, R. Gleiter and W. Krätschmer, Chem. Commun., 1999, 465 and references therein. Search PubMed.
- G. Oszlányi, G. Bortel, G. Faigel, L. Gránásy, G. M. Bendele, P. W. Stephens and L. Forró, Phys. Rev. B., 1996, 54, 11849 CrossRef CAS.
- The chemistry and physics of heterofullerenes including the aza[60]fullerene dimer (C59N)2 have been recently reviewed: J. C. Hummelen, C. Bellavia-Lund and F. Wudl, Top. Curr. Chem., 1999, 199, 93.
- J. R. Morton, K. F. Preston, P. J. Krusic, S. A. Hill and E. Wasserman, J. Am. Chem. Soc., 1992, 114, 5454 CrossRef CAS.
- S. Osawa and E. Osawa and M. Harada, J. Org. Chem., 1996, 61, 257 and references therein. Search PubMed.
- G. Schick, K.-D. Kampe and A. Hirsch, J. Chem. Soc., Chem. Commun., 1995, 2023 RSC.
- H. L. Anderson, R. Faust, Y. Rubin and F. Diederich, Angew. Chem., Int. Ed. Engl., 1994, 33, 1366 CrossRef.
- P. Timmerman, H. L. Anderson, R. Faust, J.-F. Nierengarten, T. Habicher, P. Seiler and F. Diederich, Tetrahedron, 1996, 52, 4925 CrossRef CAS.
- P. Timmerman, L. E. Witschel, F. Diederich, C. Boudon, J.-P. Gisselbrecht and M. Gross, Helv. Chim. Acta, 1996, 79, 6 CrossRef CAS.
- K. Komatsu, N. Takimoto, Y. Murata, T. S. M. Wan and T. Wong, Tetrahedron Lett., 1996, 37, 6153 CrossRef CAS.
- N. Dragoe, S. Tanibayashi, K. Nakahara, S. Nakao, H. Shimotani, L. Xiao, K. Kitazawa, Y. Achiba, K. Kikuchi and K. Nojima, Chem. Commun., 1999, 85 RSC.
- J. Osterodt and F. Vögtle, Chem. Commun., 1996, 547 RSC.
- T. S. Fabre, D. Treleaven, T. D. McCarley, C. L. Newton, R. M. Landry, M. C. Saraiva and R. M. Strongin, J. Org. Chem., 1998, 63, 3522 CrossRef CAS.
- T. Suzuki, Q. Li, K. C. Khemani and F. Wudl, J. Am. Chem. Soc., 1992, 114, 7300 CrossRef CAS.
- P. Belik, A. Gügel, J. Spickermann and K. Müllen, Angew. Chem., Int. Ed. Engl., 1993, 32, 78 CrossRef.
- A. Gügel, P. Belik, M. Walter, A. Kraus, E. Harth, M. Wagner, J. Spickermann and K. Müllen, Tetrahedron, 1996, 52, 5007 CrossRef.
- L. A. Paquette and R. J. Graham, J. Org. Chem., 1995, 60, 2958 CrossRef CAS.
- Y. Murata, N. Kato, K. Fujiwara and K. Komatsu, J. Org. Chem., 1999, 64, 3483 CrossRef CAS.
- H. Irngantinger and A. Weber, Tetrahedron Lett., 1996, 37, 4137 CrossRef.
- M. Prato and M. Maggini, Acc. Chem. Res., 1998, 31, 519 CrossRef CAS.
- A. I. de Lucas, N. Martín, L. Sánchez and C. Seoane, Tetrahedron Lett., 1996, 52, 9391 CrossRef CAS.
- N. Martín, L. Sánchez, B. Illescas and I. Pérez, Chem. Rev., 1998, 98, 2527 CrossRef CAS.
- J.-P. Bourgeois, F. Diederich, L. Echegoyen and J.-F. Nierengarten, Helv. Chim. Acta, 1998, 81, 1835 CrossRef CAS.
- S. Higashida, H. Imahori, T. Kaneda and Y. Sakata, Chem. Lett., 1998, 605 CrossRef CAS.
- D. Armspach, E. C. Constable, F. Diederich, C. E. Housecroft and J.-F. Nierengarten, Chem. Eur. J., 1998, 4, 723 CrossRef CAS.
- S. Ravaine, P. Delhaès, P. Leriche and M. Sallé, Synth. Met., 1997, 87, 93 CrossRef CAS.
- J. L. Segura and N. Martín, Tetrahedron Lett., 1999, 40, 3239 CrossRef CAS.
- R. A. J. Janssen, P. A. van Hal, J. Knol and K. Hummelen, European Conference on Organic Solar Cells (ECOS 1998)..
- T. Habicher, J.-F. Nierengarten, V. Gramlich and F. Diederich, Angew. Chem., Int. Ed. Engl., 1998, 37, 1916 CrossRef CAS.
- N. Armaroli, F. Diederich, C. O. Dietrich-Buchecker, L. Flamigni, G. Marconi, J.-F. Nierengarten and J.-P. Sauvage, Chem. Eur. J., 1998, 4, 406 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2000 |