From simple to supramolecular cytochrome P450 mimics
Martinus C. Feiters, Alan E. Rowan and Roeland J. M. Nolte
Department of Organic Chemistry, NSR Center, University of Nijmegen, Toernooiveld 1, 6525 ED, Nijmegen, The Netherlands
First published on UnassignedUnassigned2nd October 2000
Abstract
Cytochrome P450 is one of Nature’s oxidative workhorses and is utilized in a wide variety of roles, one of the most important being the detoxification of foreign bodies within the liver. As a result of its fundamental importance it has been extensively investigated, modeled and mimicked over the past 30 years, and more recently modified and mutated. During this period the complexity, beauty and activity of the biomimetic model systems developed in the laboratory have grown considerably. The synthetic analogues of the cytochrome P450 system have evolved dramatically from simple sterically hindered porphyrin models through to more complex model systems combining cavities such as cyclodextrins and utilizing the interactions between host and guest to generate substrate selectivity and stereoselectivity in product formation. More recently, researchers have tried to combine knowledge obtained from the developing field of supramolecular chemistry and from biochemistry to construct self-assembling systems that contain all the components of the natural system and even utilize molecular oxygen as the oxidant. These systems are successful in that they can achieve turnover numbers comparable to those observed for the natural system. The history of these developments and the current ‘state-of-the-art’ in construction of mimics of the natural enzyme will be presented.
 Martinus C. Feiters Martinus C. Feiters | Martin Feiters graduated in biochemistry, bio-organic chem- istry and food chemistry at the University of Utrecht. For his PhD he worked on the structure–function relationship of the enzyme lipoxygenase at the University of Utrecht and Chalmers Technical University (Gothenburg, Sweden). He subsequently did postdoctoral work in X-ray absorption spectroscopy at the Daresbury Laboratory, University of Manchester. He was appointed Associate Professor of Bio-organic Chemistry at the University of Nijmegen in 1989. |
 Alan E. Rowan Alan E. Rowan | Alan Rowan completed his PhD in physical organic chemistry in 1991 at the University of Liverpool, England. After a period of postdoctoral research at the University of Otago, Dunedin, New Zealand he returned to Europe and became an Assistant Professor at the University of Nijmegen. His scientific interests are in the design and construction of supramolecular assemblages possessing catalytic and electronic properties. |
 Roeland J. M. Nolte Roeland J. M. Nolte | Roeland Nolte received his PhD in physical organic chemistry from the University of Utrecht (1973), where he stayed and became Assistant Professor and then Associate Professor. In 1981, he was a visiting scientist at UCLA in the group of Donald J. Cram. In 1987 he moved to Nijmegen and became Professor of Organic Chemistry, and since 1994 he has also been a Professor of Supramolecular Chemistry at the Eindhoven University of Technology. His principal research interest is supramolecular chemistry, focusing on the design of catalysts and molecular materials. |
1 Introduction
Cytochrome P450 is a member of a ‘super family’ of enzymes which can be found in a wide diversity of roles throughout Nature.1 They are of fundamental importance in our everyday life due their presence within our liver where they play an important role in the detoxification of foreign bodies. Due to the numerous and fundamental roles cytochrome P450 plays, this enzyme has been extensively investigated by researchers throughout the scientific community ranging from biologists to chemists and physicists through to physicians. Yet despite this intense effort, the precise mechanism of its function is still not fully known. The wealth of information obtained, however, has enabled the (bio)organic chemist to develop simple organic analogues, which have been utilized, as both models to further understand the mechanism of enzyme action and as functional catalytic systems.Cytochrome P450 belongs to the group of enzymes known as monooxygenases which incorporate one oxygen atom of molecular oxygen into organic substrates, the other oxygen atom being converted into water [eqn. (1)].
| O2 + S + NAD(P)H + H+ → H2O + SO + NAD(P)+ | (1) |
Cytochrome P450 is catalytically active in the oxidation of a variety of biological substrates and xenobiotics and furthermore in processes such as the dehydrogenation of valproic acid and testosterone, the oxidative deformylation of aldehydes, and the dehydration of aldoximes. There are also several cytochrome P450 enzymes which do not carry out any oxidations at all but, for example, induce the isomerization of prostaglandin H2 to prostacyclin, activate allene oxides or even reduce nitric oxides (for selected examples, see Scheme 1).2
 | ||
| Scheme 1 Reactions catalysed by cytochrome P450 and related enzymes. | ||
The primary driving force for the extensive study of this enzyme is to further investigate the mechanistic behaviour of the natural system and to subsequently construct a new generation of organic oxidative catalysts.
2 Structure and activity of cytochrome P450
The mammalian cytochrome P450 enzymes present in eukaryotic organisms reside in the endoplasmatic reticulum, and will be found in the microsomes upon cell fractionation. They differ from cytochrome P450 found in microorganisms in that they are bound within the membrane. The hydrophobic nature of the enzymes has severely hindered their crystallization due to the tendency of the protein to aggregate rather than form single crystals. Only very recently have a number of crystal structures of cytochrome P450 from soluble microorganisms been obtained, the very first being that of the enzyme from Pseudomonas putida which selectively hydroxylates the substrate camphor in the 5-exo position (Fig. 1).3 Later other structures from bacterial sources were reported which have become important as a basis for site-directed mutagenesis studies and have also been invaluable in enabling the construction of 3-dimensional structures of cytochrome P450s for which only the amino acid sequences are known.4 In a very recent modeling study by Dai in 1998, the part of the protein that inserts in the membrane has been identified, and the membrane interaction studied.5 There is strong evidence that in the case of microsomal cytochrome P450 enzymes the presence of certain membrane components like phospholipids play a significant role in their catalytic activity. | ||
| Fig. 1 X-Ray crystal structure of the cytochrome P450 from Pseudomonas putidas which selectively hydroxylates camphor. The iron protoporphyrin IX, the coordinated cysteine ligand and the camphor substrate are highlighted. | ||
Cytochrome P450 contains a binding site and next to it a catalytically active iron protoporphyrin IX molecule (Fig. 1). Together with a number of other proteins, e.g. chloroperoxidase, cytochrome P450 belongs to the group of heme thiolate proteins, which feature a proximal thiolate ligand from the amino acid cysteine. It is in fact this ligand that is responsible for the remarkable red shift of the heme absorption (Soret) band from 420 to 450 nm upon CO complexation to the reduced enzyme, which gave cytochrome P450 its name.
A feature that distinguishes cytochrome P450 and chloroperoxidase (which have no further similarity at either the primary or tertiary structure level)6 from other heme proteins is the fact that the proximal ligand is at the positive end of an α-helix creating an electropositive environment and diminishing somewhat the effect of the negative charge on the thiolate. As a consequence of this feature the soft and electron donating nature of the thiolate is thought to contribute to the splitting of the oxygen–oxygen bond of molecular oxygen (vide infra).7 It is believed that the ligand confers a radical nature to the active oxidant and enhances its electrophilicity in the catalytically active species. This effect is of great relevance to the mechanism of action of cytochrome P450.
3 Mechanism of action of cytochrome P450
The native enzyme when isolated was found to contain a low-spin FeIII porphyrin, which is a little surprising since the thiolate ligand is hardly a sufficiently strong ligand to bring about the change from the expected high-spin. It was initially thought that water which fills the substrate binding pocket plays a role in the realization of this low-spin state.3 As a result of extensive computational modeling, however, it is now agreed that the axial thiolate ligand alone is responsible for the fact that native cytochrome P450 is low-spin.8The catalytic cycle begins with the displacement of the water cluster from the binding site by a substrate molecule. This substrate binding changes the redox potential of the iron center or induces a conformational change that allows more effective interaction of the enzyme with the electron donor. In microsomal systems the reductant is a flavin-containing enzyme called cytochrome P450 reductase that derives its electrons from the oxidation of NADPH (see Fig. 2). It is important to realize that while NADPH delivers its electrons in pairs, they are passed on sequentially one by one to cytochrome P450. The first electron (see Figs. 2 and 3) is delivered only after substrate binding and changes the state of the iron to low-spin FeII. Molecular oxygen is then bound to produce a dioxygen adduct; the accepted electron is transferred from the iron center to oxygen, giving a FeIII–peroxo complex. This FeIII peroxo species can accept another electron from its physiological redox partner and then be quickly protonated to the FeIII hydroperoxo form. An additional proton is accepted and a molecule of water is split off, resulting in a reactive intermediate containing an electrophilic oxygen atom which is responsible for the oxygen transfer to organic substrates, e.g. alkanes (Fig. 3). Studies on model compounds have shown that the thiolate ligand enhances the rate of the O–O cleavage in these complexes and promotes heterolytic splitting rather than homolytic O–O splitting.
 | ||
| Fig. 2 Schematic picture of the cytochrome P450 enzyme system. | ||
 | ||
| Fig. 3 Catalytic cycle for cytochrome P450 monooxygenations. | ||
The reactive intermediate has the same oxidation state as compound I in
the catalytic cycle of horseradish peroxidase, and is formally a
O![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) FeVP species, but is better described as a ferryl
OFeIVP+· species with a
radical cation on the porphyrin ring, although currently there is only
indirect evidence for this species.9 More
recent studies indicate that the S of the complexing thiol ligand also
possesses some radical character.10
Numerous studies have been carried out in order to pinpoint the precise
structure of the reactive species, in particular their spin states. These
spin states are important because of the concept of two-state
reactivity11 that has been developed
recently in order to account for the findings of the so-called
‘radical clock’ experiments. These experiments show that
hydroxylations by cytochrome P450 appear to proceed by free radicals but
that the product distribution is not in line with the expected radical
lifetimes.12 Like dioxygen, the porphyrin
radical cation–FeIVO complex is proposed to have a
high-spin ground state which should lead to a low reactivity in reactions,
like C–H abstraction, which involve low-spin transition states or
products.11 Such a low reactivity is not
observed. It is suggested, therefore, that one of the possible pathways
from the high-spin reactants to the low-spin products involves a spin
inversion by which a low-spin transition state of relatively low energy can
be reached. In this two-state reactivity mechanism, reactants are converted
into products through a transition state of different spins, as opposed to
a single-state reactivity, in which the spin state stays the same
throughout the reaction. It has very recently been demonstrated by Shaik
et al. that the contradictory data observed so far for the
cytochrome P450 mechanism can be reconciled within this two-state
scenario.11 It is clear from the discussion
above that there is still considerably more research required before the
precise mechanism of action of the cytochrome P450 will be conclusively
understood.
FeVP species, but is better described as a ferryl
OFeIVP+· species with a
radical cation on the porphyrin ring, although currently there is only
indirect evidence for this species.9 More
recent studies indicate that the S of the complexing thiol ligand also
possesses some radical character.10
Numerous studies have been carried out in order to pinpoint the precise
structure of the reactive species, in particular their spin states. These
spin states are important because of the concept of two-state
reactivity11 that has been developed
recently in order to account for the findings of the so-called
‘radical clock’ experiments. These experiments show that
hydroxylations by cytochrome P450 appear to proceed by free radicals but
that the product distribution is not in line with the expected radical
lifetimes.12 Like dioxygen, the porphyrin
radical cation–FeIVO complex is proposed to have a
high-spin ground state which should lead to a low reactivity in reactions,
like C–H abstraction, which involve low-spin transition states or
products.11 Such a low reactivity is not
observed. It is suggested, therefore, that one of the possible pathways
from the high-spin reactants to the low-spin products involves a spin
inversion by which a low-spin transition state of relatively low energy can
be reached. In this two-state reactivity mechanism, reactants are converted
into products through a transition state of different spins, as opposed to
a single-state reactivity, in which the spin state stays the same
throughout the reaction. It has very recently been demonstrated by Shaik
et al. that the contradictory data observed so far for the
cytochrome P450 mechanism can be reconciled within this two-state
scenario.11 It is clear from the discussion
above that there is still considerably more research required before the
precise mechanism of action of the cytochrome P450 will be conclusively
understood.
In addition to alkane hydroxylation, cytochrome P450 enzymes also
oxidize alkenes to form epoxides and carry out the dealkylation of amines.
The oxidation of alkenes is a reaction of great interest to the organic
chemist. Detailed studies on the interaction of the porphyrin FeO
species with alkenes by Gross have led to the proposal that the first
intermediate on the reaction pathway of epoxide formation is an ion pair,
formed by electron transfer from the alkene to the FeO complex,
whereas studies by Bruice indicate that the first intermediate is a charge
transfer complex.13 Axial ligands that
stabilize high metal oxidation states are more likely to push the
interconversion equilibrium to the right[eqn. (2)] thereby facilitating oxygen
transfer.13
| FeIV(O)Porph·
⇌
FeV(O)Porph | (2) |
4 Synthetic model systems
A wide variety of approaches has been developed to design and construct synthetic model systems of the cytochrome P450 family. In addition to models synthesized to mimic specific aspects of the action of the natural system that aid in the understanding of the mechanism, working catalytic systems have been designed using the information gained from the aforementioned studies as a blueprint. One primary reason why cytochrome P450 has attracted so many organic chemists is that there is no similar hydroxylation reaction of hydrocarbons known in organic chemistry.One can construct a catalytic system using another protein or enzyme found in nature. An example of such a catalyst is the so-called microperoxidase that is obtained by limited proteolysis of cytochrome c. This biomolecule contains a proximal imidazole ligand and catalyses cytochrome P450-type reactions like aromatic hydroxylation and thioether oxidation with incorporation of oxygen from the oxidant.15 This elegant approach is, however, not ideal since it does not allow the catalyst to be easily modified and its properties manipulated. In the case of more simple model systems the catalyst can be much more easily controlled by altering the proximal ligand, the electron state of the porphyrin, the nature of the binding environment (chiral, hydrophobic–hydrophilic, polar–nonpolar) and a wide variety of other parameters.
It has been found that the combination of molecular oxygen and a cofactor in the enzyme is complex, since regeneration in vitro is difficult and also the reducing agent can compete with the substrate for the active oxidant. This problem can be circumvented by using so-called ‘single oxygen donors’, e.g. sodium periodate, iodosobenzene, hypochlorite, hydrogen peroxide, a peroxy acid and tert-butyl hydroperoxide.16–20 This has led to the development of synthetic cytochrome P450 models in which the (difficult to achieve) reductive activation of molecular oxygen is by-passed by this so-called ‘peroxide shunt’ (Fig. 3).
Another problem encountered in model systems is the extreme sensitivity of the combination of the thiolate group and FeII to air and light, and the tendency of thiolate ligands to give bis-axial complexes.20
One approach to overcome this problem is to take aromatic thiols as ligands which have a reduced sensitivity to oxidation21 or to use doubly strapped porphyrins in which one or two thiolate ligands are held rigidly in one strap.22 Using such model systems it has been possible to make a detailed assessment of the importance of the thiolate ligand for the properties and action of the enzyme.23 The aforementioned hypothesis, that the thiolate ligand helps confer radical character on the oxygen received support from a comparative study of kinetic isotope effects and 18O-incorporation from 18O-enriched oxidants in the dealkylation of aryl alkyl ethers using rat liver microsomes and a porphyrin with a covalently attached thiolate ligand.23
Hydrogen bonding networks involving the proximal thiolate ligand have been identified in cytochrome P450 and in the related enzyme chloroperoxidase. This feature has also been mimicked, and a model system in which the thiolate group accepts H-bonds from neighbouring amide groups was found to have considerable stability towards degradation by oxygen and moisture.24,25
Although thiols are the natural proximal ligands of cytochrome P450, and have been demonstrated to be more effective in model catalysts than nitrogen-based ligands, the aforementioned problems with the instability of thiol-ligated metal–oxo complexes (along with a purely human problem, the unpleasant aroma of such systems), have led many scientists to resort to the use of simple iron(III) porphyrins with a halide axial ligand and/or porphryrin systems with imidazole or pyridine ligands.
The very first model systems using nitrogen base ligands were simple functional iron porphyrins, in which particular emphasis was laid on creating steric bulk around the porphyrin in order to prohibit the most important reactions that cause instability of heme–oxygen complexes to occur, viz. the dimerization with free heme, and porphyrin ligand oxidation. With subsequently developed models,26 using capped porphyrins (Fig. 4) an attempt was made to avoid the binding of the nitrogen ligands to both axial positions of the iron center which would inhibit the binding of molecular oxygen or the single oxygen donor.27
 | ||
| Fig. 4 Model cytochrome P450 systems: simple pyridine capped porphyrin model (1), ‘picnic basket’ porphyrin model developed by Collman (2), and the ‘basket-handle’ porphyrin of Mansuy (3). Chiral porphyrin cytochrome P450 models sytems: tetra-diethenoctahydrotetralin functionalized porphyrin (4) and binap-TCP systems (5a eclipsed and 5b staggered). | ||
More recently model systems have focused on the development of functionalised porphyrins for regio- and stereoselective oxidation reactions.28 Examples are the capped Mn porphyrins, which in combination with pyridine as an axial ligand give a high selectivity for the formation of the cis-product in the epoxidation of (Z)-stilbene with NaOCl (Fig. 4, 1).28 Substrate selectivity has been achieved with the so-called ‘picnic basket porphyrins’ which contain a rigid cavity (Fig. 4, 2). The Mn derivatives of these porphyrins showed a preference for the epoxidation of (Z)-oct-2-ene over cyclooctene in competition experiments.28 There are numerous examples of enantioselective oxo-transfer by substituted porphyrins, e.g. with chirality in the strap, as in the ‘basket-handle’ porphyrin (Fig. 4, 3),2 or with a bulky chiral group at the periphery of the porphyrin ring, as in the ‘chiral wall’ porphyrin,28 or with diethenoctahydrotetralin substituents (Fig. 4, 4).28 Very high enantioselectivity is found for the so-called ‘twin coronet porphyrins’. In these porphyrins the phenyls are linked by binapthalenes (Fig. 4, 5a eclipsed, 5b staggered) or bitetralins. In most cases the bitet-TCP’s gave better ee’s, in particular for the Fe derivative of the eclipsed ‘bitet-TCP’ ee’s of 96% were obtained for the product of the epoxidation of 3,5-dinitrostyrene.28 A Mn derivative of the aforementioned ‘picnic basket porphyrin’ with (S,S)-threitol acetal moieties in the handle catalyses the epoxidation of (Z)-α-methylstyrene and 1,2-dihydronaphthalene with ee values of 77–88%.28
With the advent of supramolecular chemistry, cytochrome P450 mimics containing a porphyrin attached to a cavitand like a cyclodextrin or a cyclophane have become increasingly popular. A biomimetic catalyst for alkene epoxidation, composed of two cyclodextrin cavities, viz. a cyclodextrin with an alkanethiolate ligand and a cyclodextrin capped with a porphyrin, linked on their primary sides by a flavin unit has been prepared by Breslow’s group.29 This group, and the groups of Ogoshi and Hennig have synthesized a wide variety of cyclodextrin–porphyrin model systems of cytochrome P450 (Fig. 5, 1–4).30–32 Cyclodextrin-sandwiched porphyrins were found to be equally good catalysts for the epoxidation of 2,3-dimethylbut-2-ene, which does not bind in the cyclodextrin cavity, and even better catalysts for substrates that do bind within the cavity, such as cyclohexene (Fig. 5, 2).31 The group of Breslow has attached several cyclodextrin cavities to one porphyrin molecule in order to improve both the substrate binding properties and the substrate selectivity of the model system. The porphyrin with four cyclodextrins (Fig. 5, 3) is able to bind cholesterol and alkene derivatives in a specific orientation with respect to the central porphyrin, resulting in a catalyst that displays site-specific and stereoselective hydroxylation reactions. In the case of functionalized alkenes, oxidation with iodosobenzene generates the epoxides with turnover numbers in the range of 650.30
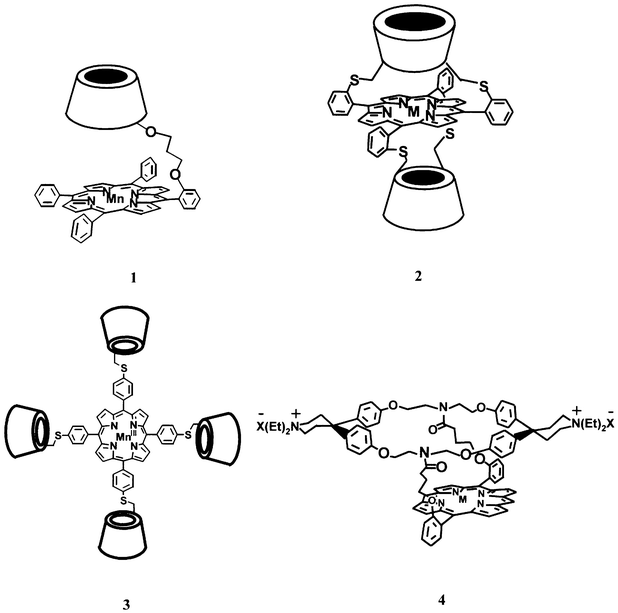 | ||
| Fig. 5 Supramolecular cytochrome P450 model systems containing cyclodextrin and cyclophane cavities developed by Ogashi (1), Hennig (2), Breslow (3) and Diederich (4). | ||
In an alternative approach the group of Diederich has attached an Fe porphyrin to an azacyclophane host that contains quaternized diethylcyclohexylamine rings to make the system water-soluble (Fig. 5, 4). This supramolecular molecule was found to catalyse the transfer of oxygen from iodosobenzene to acenaphthylene in 2,2,2-trifluoroethanol, yielding acenaphthylen-1-one. Porphyrins incorporated in cyclophane rings have also been synthesized.33
5 Catalytic antibodies
An interesting approach for the development of new catalysts is to use ‘catalytic antibodies’. The principle is based on the idea that Pauling proposed in 1948, that an enzyme is an efficient catalyst for a particular reaction because it stabilizes the transition state of that reaction.34 If a stable analogue for a transition state of a reaction can be designed and prepared and this can be applied as a hapten to produce antibodies, the latter are likely to interact with this transition state in such a way that they will catalyse the reaction. In the field of cytochrome P450, antibodies catalysing the oxo-transfer step have been developed.Various examples of antibodies raised against porphyrins have been reported35 and catalytic activities such as porphyrin metallation,36 peroxidase activity, i.e. degradation of pyrogallol and other substrates, have been observed.37 Relatively simple porphyrins, like the Sn(OH)2 derivative of a meso-tetrakis-(4-carboxyvinylphenyl) porphyrin or its FeIII μ-oxo dimer, were found to give circular dichroism spectra when combined with the antibodies that were raised against them, indicating that they are bound within the (chiral) protein environment.38 The activity of such antibody–porphyrin assemblies in the oxo transfer from iodosobenzene to styrene was tested in a number of biphasic systems,38e.g. reverse micelles in dichloromethane, microemulsions, and solid suspensions in organic solvents. Only in the last case was there observed some rate enhancement (30–60%) relative to the reaction in the absence of the antibody. The background reaction involving the non-complexed porphyrin must be relatively strong, as is also reflected by the low ee values (10%) that were observed. Very recently, it was demonstrated that far more efficient catalytic antibodies can be obtained by applying dinaphthoxy tin porphyrin derivatives as the hapten.39 When recombined with the corresponding RuII porphyrin, they were found to catalyse oxo transfer from iodosobenzene to a variety of sulfides to yield chiral sulfoxides. The highest rate was obtained with p-methoxythioanisole (kcat/kuncat 6.5; ee of (S)-product 27%), and the highest ee with unsubstituted thioanisole (kcat/kuncat 2.8; ee of (S)-product 43%). The dinaphthoxy substituted tin porphyrin can be considered to be a transition state analogue for both the step of oxygen transfer from iodosobenzene to the porphyrin and the subsequent oxygen transfer from the porphyrin to the sulfide. The observed ee indicates that the transfer of oxygen from the Ru-porphyrin to the sulfide presumably takes place in the chiral environment of the antibody. Interestingly, in some examples the oxygen transfer catalysis by the antibodies proceeded even without the presence of porphyrins, but this will not be discussed in this review.40–42
6 Self-assembled model systems
The first report of reductive activation of molecular oxygen in a model system was by the group of Tabushi in 1979.43 They demonstrated that cyclohexanol can be formed from cyclohexene and molecular oxygen using manganese(III) tetraphenylporphyrin chloride (MnTPPCl) as a catalyst and NaBH4 as a reducing agent. Similar reactions have since been reported for a variety of other metallated porphyrins, i.e. Rh, Co and Fe porphyrins. Using molecular hydrogen in combination with colloidal platinum for the reduction, and imidazole as an axial ligand, the major product of the MnTPP-catalysed oxidation of cyclohexene with molecular oxygen was cyclohexene epoxide.44 Several supramolecular self-assembled systems have been described in which the oxygen is derived from single oxygen donors or from molecular oxygen and a co-reductant.In the cases where not just oxygen transfer but complete reductive activation of molecular oxygen by cytochrome P450 is modeled, the problem is to find a suitable reductant and to control the flow of electrons and protons in such a way that complete reduction of oxygen to water, without incorporation of an oxygen atom into an organic substrate, is avoided. Several approaches have been followed to overcome this difficult problem. Closely mimicking nature, flavin molecules have been covalently linked to an Fe- or Mn-porphyrin and have been combined with an imidazole-substituted nicotinamide.29 Alternative reductants such as NaBH4, Zn, and Zn(Hg) have all been used. Very recently, an azobenzene reductant in combination with a viologen mediator has been applied as well.44 Advantage has also been taken of the possibility to reduce the porphyrins with the help of light, either by initializing a process in which a ligand radical is dissociated from the porphyrin which acts as an initiator of a radical chain auto-oxidation reaction, or by using a charged semiconductor like solid TiO2 which supplies the necessary electrons.
An interesting aspect of cytochrome P450 is the fact that it is a membrane-bound enzyme, and that phospholipids stimulate the transfer of electrons to the isolated enzyme and enhance its affinity for the substrate. This feature has stimulated a number of groups to develop cytochrome P450 mimics in which a hydrophobic porphyrin is incorporated in the bilayer of a vesicle membrane.
Sorokin in the early 1980’s constructed a system in which the hydrophobic porphyrin manganese(III) tetrahexadecylphenylporphyrin chloride was incorporated within dimizistoylphosphatidylcholine (DMPC) vesicles.45 They studied oxidation reactions with iodosobenzene as an oxidant. The oxidation of hydrocarbons was remarkably selective, with hexan-2-ol from hexane, and cyclohexanol from cyclohexane as the major and exclusive products, respectively. Cyclohexene gave the epoxide and the allylalcohol in relatively high yields, viz. 40 and 30% based on the iodosobenzene consumed. By incorporating a membrane-spanning FeIIITPP chloride derivative containing four steroid groups, viz. tetrakis(o-cholenylamidophenyl)porphyrin (ChPP), in DMPC or dipalmitoylphosphatidylcholine (DPPC) bilayers, regioselectivity in the epoxidation of steroids and polyunsaturated fatty acids has been achieved by Groves and co-workers using iodosobenzene as the oxidant.46 In the case of steroids, the double bonds farthest away from the hydroxy group were selectively epoxidized in 20–30% yields, e.g. the 24,25 double bond in desmosterol and the 24,28 double bond in fucosterol. This contrasts with the epoxidation of these substrates by FeIIITPPCl in methylene chloride which showed a significant 3–4 fold preference for the epoxidation of the 5,6 double bond in both cases.46 In another study, selective hydroxylation of cholesterol at the hydrophobic 25-position was catalysed by MnIIItetrakis(o-cholenylamidophenyl)porphyrin in dilauroylphosphatidylcholine (DLPC) vesicles with molecular oxygen as the oxidant and ascorbic acid as the reducing agent, or with sodium periodate as the single oxygen donor. Interestingly, with cyclooctane and ethylbenzene as the substrates, the ketones cyclooctanone and acetophenone were obtained, respectively, when the aerobic system was applied46 whereas the single oxygen donor gave mixtures in which the alcohol predominated. The group of Groves has developed a more complex self-assembled system in which the reductive activation of molecular oxygen by MnIII ChPP in DMPC bilayers is realized with the help of a membrane-associated enzyme, viz. the flavoprotein pyruvate oxidase. In this system, electrons are derived from the oxidative decarboxylation of pyruvic acid and carried to the porphyrin by an amphiphilic flavin. The enzyme is known to bind to phospholipid vesicles when the required cofactors and the substrate are present, presumably due to a conformational change in the protein which exposes a membrane-binding hydrophobic segment. After 15 hours at 30 °C, the system had produced 20 per mol acetophenone per mol Mn ChPP from ethylbenzene. This hybrid enzyme–synthetic model system is referred to as a ‘biocompatible catalyst’ (Fig. 6).47
 | ||
| Fig. 6 Schematic representation of the biocompatible catalytic system developed by the group of Groves, FAD = flavoprotein pyruvate oxidase. | ||
In an alternative system, the group of Nolte incorporated a manganese porphyrin in the bilayers of vesicles of a polymerizable isocyanide analogue of dihexadecyldimethylammonium bromide (DHDAB).48 Within the vesicles colloidal platinum was encapsulated, together with methylene blue as an electron donor and N-methylimidazole as an axial ligand for the porphyrin. This system catalyses at 20.0 °C under an atmosphere of hydrogen and oxygen gases (1∶1) the reductive activation of oxygen to form epoxides (predominantly detected as the corresponding diols) of both water-soluble (2,5-dihydrofuran) and water-insoluble (styrene) alkenes, with turnover numbers of 8 and 1.3 mol product per mol porphyrin per hour, respectively (Fig. 7).
 | ||
| Fig. 7 Vesicle cytochrome P450 model system developed by the group of Nolte (top); Freeze fracture electron micrographs of the unpolymerized (A) and polymerized (B) vesicles which hold the components of the model system (bottom). | ||
This catalytic system incorporates all the features of the natural enzyme system, viz. a metalloporphyrin, an axial ligand, an electron donor, an electron carrier, and a membrane which holds all components. As the molecular hydrogen–platinum reduction system is inefficient, and the combination of hydrogen and oxygen gases (detonating gas) dangerous, alternative reducing agents have been investigated, viz. the RhIII–formate system. It was found that RhIII(pentamethylcyclopentadienyl) (2,2′-bipyridyl) bis-chloride in combination with formate ions can be applied as a reductant and at the same time as a phase-transfer catalyst in a two-phase catalytic system for the reductive activation of oxygen, viz. with the formate in the aqueous layer, and MnTPP Cl and the alkene substrate in the 1,1,1-trichloroethane layer.49 The turnover numbers are higher in the presence of benzoic acid anhydride, which is proposed to assist in the cleavage of the oxygen–oxygen bond in molecular oxygen after coordination of the latter to Mn TPP. At 40 °C, α-pinene is selectively converted into the epoxide (30 mol per mol porphyrin per hour), and nerol to a mixture of 2,3- and 6,7-epoxides in the ratio 1∶2 (18 mol per mol porphyrin per hour). Recently, manganese tetrakis(2,6-dichlorophenyl)porphyrin acetate (MnTDCPP acetate) was combined with an amphiphilic 2,2′-bipyridyl Rh (pentamethylcyclopentadienyl) complex (Scheme 2, 1) and N-methylimidazole in both dihexadecyl phosphate (DHP) and dioctadecylammonium chloride (DODAC) vesicles.50
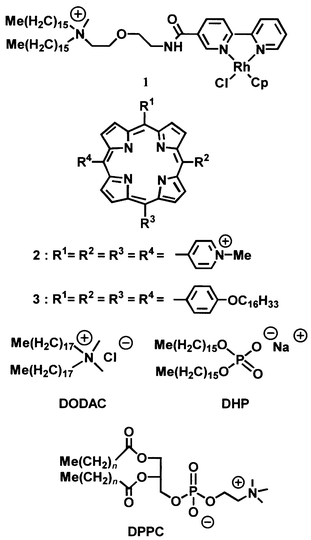 | ||
| Scheme 2 Chart of the rhodium complex (1), porphyrins (2, 3) and amphiphile (DODAC, DHP, DPPC) used in the Nolte self-assembled cytochrome P450 mimic. | ||
The reduction of MnIII TDCPP by formate under argon is faster in DODAC than in DHP, because of the accumulation of formate ions on the positively charged surface of the former vesicles. Nevertheless, with styrene as substrate, the DHP vesicles were a more efficient support for the epoxidation catalysts in the presence of oxygen (turnover 60 mol per mol porphyrin per hour at 70 °C), than DODAC vesicles presumably because at the Rh∶Mn ratio used (1∶1 mol/mol), the availability of the protons required for the epoxidation is rate limiting. Comparison of the vesicle with the two-phase system showed a higher turnover number for the former system (360 vs. 30 mol per mol porphyrin per hour for α-pinene at 70 and 40 °C, respectively), a higher stability of the porphyrin, and no interference of the imidazole with the reduction of the Rh complex by formate.
A detailed study was carried out to obtain information about the precise behaviour of the catalyst and cofactor in the last mentioned cytochrome P450 mimic.51 To this end the location, aggregation and orientation of a number of tetraarylporphyrins in vesicle bilayers of both positively and negatively charged amphiphiles, viz. DODAC and DHP, were investigated in detail. It was concluded that in DODAC bilayers charged porphyrins (Scheme 2, 2) form face-to-face aggregates, whereas hydrophobic ones (Scheme 2, 3), yieldedge-to-edge aggregates and that the monomeric porphyrins have an orientation perpendicular to the normal of the DODAC bilayer.51
The efficiency of the reduction of the amphiphilic Rh co-catalyst
N-(2-(2-(2,2′-bipyridyl-5-ylcarboxamido)ethoxy)ethyl)-N
,N-dihexadecyl-N-methylammonium) rhodium dichloride
(Scheme 2, AmphRh, 1) by
formate in vesicle bilayers of various types of amphiphiles, viz.
cationic (DODAC), anionic (DHP), and zwitterionic (DPPC), and at various pH
values was investigated. At pH values where the reduced Rh complex is
expected to be present as RhI only, the rate of the reduction of
the amphiphilic Rh complex by formate increased in the series
DPPC<DHP<DODAC, in line with the expected higher concentration of
formate ions at the surface of the cationic vesicles52 (Table 1).
The reduction rates of the porphyrins incorporated in the vesicle bilayers
catalysed by the amphiphilic Rh complex increased in the same order as
mentioned above (DPPC<DHP<DODAC) because formation of the
Rh–formate complex is the rate-determining step in this reduction.
However, when the rates of epoxidation of styrene in this cytochrome P450
mimic (Fig. 8) were studied at pH 7, the
relative rates were found to be reversed, with almost total absence of
product formation in the case of DODAC: DODAC<<DPPC<DHP.
Apparently, for epoxidation to occur, an efficient supply of protons to the
vesicle surface is important, probably for the step in which the
MnII–O2 complex breaks down into the active
epoxidizing MnVO species and water. With α-pinene
as the substrate, a turnover number of 360 was observed using DHP vesicles
above the phase transition temperature. This is comparable to the turnover
numbers of cytochrome P450 itself at room temperature.
| AmphRh | MnIII TDCPP Cl | |||
|---|---|---|---|---|
| Surfactant | pKaobs | Reductionak1/ms−1 | Reductionbk0/nmol l−1 s−1 | Epoxidation turnover no. h−1c |
| a First-order rate constant for the reduction by formate ions.b Zero-order rate constant for the reduction by AmphRh and formate ions.c Epoxidation of styrene by MnTDCPPCl, AmpRh, formate and O2.d Reduction rate measured for MnIII-5-(1-methyl-4-pyridyl)-10,15,20-tris(4-hexadecyloxyphenyl )porphyrin monotosylate.51 | ||||
| DHP (72–75 °C) | 8.6 | 10 | 60 | 55 |
| DPPC (47–48 °C) | 5.7 | 3.2 | 12.5d | 4 |
| DODAC (72–75 °C) | 5.0 | 25 | >100 | 0 |
 | ||
| Fig. 8 Schematic represention of the rhodium–porphyrin vesicle system developed by Nolte et al. (top, middle). Change in absorbance at 435 nm (MnII) versus time under the conditions at which the oscillating reduction of manganese(III) porphyrin takes place (bottom). | ||
An intriguing aspect of the self-assembled system depicted in Fig. 8 is that at specific conditions an oscillatory behaviour was observed.52 At 48 °C and a formate concentration of 0.25M, no reduction of MnIII porphyrin was observed at [Rh]/[Mn] ratios lower than 10. At [Rh]/[Mn] ratios higher than 10, reduction took place. At a ratio of exactly 10, oscillations in the concentration of the MnII species were detected. After an initial induction period of 30 minutes the MnII species was formed which was then converted back to the MnIII species by oxidation with the O2 present. After 50 minutes an oscillating reduction of MnIII occurred. This process was found to be exceptionally sensitive to slight changes in the temperature and the [Rh]/[Mn] ratio. At a [Rh]/[Mn] ratio < 10 the reoxidation of the MnII is faster than the reduction. At a [Rh]/[Mn] ratio > 10 the situation is reversed. At a ratio of 10 the manganese shuttles between the 2+ and 3+ states. UV–Vis studies suggested that the MnII porphyrin after reduction moves toward the middle of the bilayer, which has a less polar environment. This unique behaviour closely resembles that seen for the natural enzyme system horseradish peroxidase.
7 Dendrimers
An alternative approach to positioning porphyrins as mimics of cytochrome P450 within a hydrophobic environment is the use of dendrimers. In the past few years there have been numerous efforts to encapsulate metalloporphyrins into the interior core of dendritic polymers.53 Such systems have been shown to display interesting photophysical and electrochemical properties, as well as oxygen binding and regioselective catalysis. The group of Aida were the first to show that molecular oxygen can be readily bound to an iron porphyrin covalently encapsulated within an aryl ether dendrimer (Fig. 9, compound 1).53 It was later shown that in a similar system the oxygen can be activated by laser stimulation of the dendritic tails.53 The precise mechanism by which the oxygen is activated is not yet understood, but this approach has interesting possibilities. | ||
| Fig. 9 Dendritic cytochrome P450 model system of Aida (top). Self-assembling dendritic model system developed by Nolte et al. | ||
In addition to oxygen binding, it has also been demonstrated that by modifying the dendritic system using different generations of dendritic tails, complexation of substrates to the central porphyrin can be shape-selective. Although in its infancy, the use of dendrite–porphyrin systems has a great potential for the development of cytochrome P450-like synthetic catalysts. One slight drawback to this approach is that the dentritic tails are covalently attached to the porphyrin. The group of Nolte has developed an alternative approach in which the dendritic tails and the central porphyrin are self-assembled to give a model of the cytochrome P450 system.54 Using a tetrakis(3,5-dihydroxyphenyl)porphyrin (Fig. 9, compound 2) it was demonstrated that upon mixing with a ‘clip’-shaped diphenylglycoluril host (Fig. 9, compound 3), a 4∶1 complex was formed in which the porphyrin is completely encapsulated within the hydrophobic tails (Fig. 9, compound 4). Although to date no catalysis has been carried out with this system, the electrochemical properties of this assembly mimic more closely those of the natural system than that of a free porphyrin.54
8 Conclusion
The synthetic analogues of the cytochrome P450 system have evolved dramatically in the past twenty years from simple sterically hindered porphyrin models through to more complex model systems combining cavities and utilizing the interactions between host and guest to generate the substrate selectivity and stereoselectivity in product formation. More recently, researchers have tried to combine the knowledge obtained from the developing field of supramolecular chemistry and that obtained from biochemistry to construct self-assembling systems that contain all the components of the natural system and even utilize molecular oxygen as the oxidant. These systems are successful in that they can achieve turnover numbers comparable to those observed for the natural system. The question is where now lies the next challenge in mimicking cytochrome P450? Almost all the model systems described, aside from the exciting field of catalytic antibodies, still lack the selectivity in substrate and the stereoselectivity of the product of some cytochrome P450 enzymes found in nature. The ultimate goal is to combine the selectivity of the cavitand systems with the knowledge obtained from supramolecular chemistry and biochemistry, to design model systems which self-assemble and self-organise themselves into highly active and selective catalysts, not only mimicking the activity and selectivity of the natural system but also significantly improving on it.9 Acknowledgements
The authors thank the chemical branch of the Dutch Research Council, for support, and Dr J. H. van Esch and Dr A. P. H. J. Schenning, and the undergraduate students D. H. W. Hubert and J. H. Lutje Spelberg, for carrying out parts of the research described in this review.10 References
- J. T. Groves and Y. Z. Han, in ‘Cytochrome P450. Structure, Mechanism and Biochemistry’, ed. P. R. Ortiz de Montellano, Plenum Press, New York, 1995, pp 3–48 Search PubMed; J. M. Mayer, in Biomimetic Oxidations, ed. B. Meunier, ICP Publishers, 2000, in press. Search PubMed.
- D. Mansuy, Pure Appl. Chem., 1994, 66, 737–744 CAS; D. Mansuy and J.-P. Renaud, in Cytochrome P450: Structure, Mechanism, and Biochemistry, ed. P. R. Ortiz de Montellano, 2nd edn., Plenum Press, New York, 1995, pp. 537–574. Search PubMed.
- T. L. Poulos, J. Biol. Inorg. Chem., 1996, 1, 356–359 CrossRef CAS; T. L. Poulos, B. C. Finzel and A. J. Howard, Biochemistry, 1986, 25, 5314–5322 CrossRef CAS; T. L. Poulos, B. C. Finzel and A. J. Howard, J. Mol. Biol., 1987, 195, 687–700 CAS.
- D. F. V. Lewis, Xenobiotica, 1998, 28, 617–661 CrossRef CAS; Y. T. Chang, O. B. Stiffelman, I. A. Vakser, G. H. Loew, A. Bridges and L. Waskell, Protein Eng., 1997, 10, 119–129 CrossRef CAS.
- R. Dai, M. R. Pincus and F. K. Friedman, J. Protein Chem., 1998, 17, 121–129 CrossRef CAS; R. Dai, S. Zhai, X. Wei, M. R. Pincus, M. R. Vestal and F. K. Friedman, J. Protein Chem., 1998, 17, 643–650 CrossRef CAS.
- M. Sundaramoorthy, J. Terner and T. L. Poulos, Structure, 1995, 3, 1367–1377 CrossRef CAS.
- J. H. Dawson, Science, 1988, 240, 433–439 CAS.
- D. Harris, G. Loew and L. Waskell, J. Am. Chem. Soc., 1998, 120, 4308–4318 CrossRef CAS; M. T. Green, J. Am. Chem. Soc., 1998, 120, 10772–10773 CrossRef CAS.
- I. Schlichting, J. Berendzen, K. Chu, A. M. Stock, S. A. Maves, D. E. Benson, R. M. Sweet, D. Ringe, G. A. Petsko and S. G. Sligar, Science, 2000, 287, 1615–1621 CrossRef CAS; J. T. Groves, R. C. Haushalter, M. Nakamura, T. E. Nemo and B. J. Evans, J. Am. Chem. Soc., 1981, 103, 2884–2888 CrossRef CAS; R. Rutter, L. P. Hager, H. Dhonau, M. Hendrich, M. Valentine and P. Debrunner, Biochemistry, 1984, 23, 6809–6816 CrossRef CAS; C. E. Schultz, R. Rutter, J. T. Sage, P. G. Debrunner and L. P. Hager, Biochemistry, 1984, 23, 4743–4754 CrossRef.
- M. T. Green, J. Am. Chem. Soc., 1999, 121, 7939–7940 CrossRef CAS.
- S. Shaik, M. Filatov, D. Schröder and H. Schwarz, Chem. Eur. J., 1998, 4, 193–199 CrossRef CAS; M. Filatov, N. Harris and S. Shaik, J. Chem. Soc., Perkin Trans. 2, 1999, 399–410 RSC; S. Shaik, M. Filatov, D. Schröder and H. Schwarz, Angew. Chem., Int. Ed., 2000, in press. Search PubMed.
- S.-Y. Choi, P. E. Eaton, P. F. Hollenberg, K. E. Liu, S. J. Lippard, M. Newcomb, D. A. Putt, S. P. Upadhyaya and Y. Xiong, J. Am. Chem. Soc., 1996, 118, 6547–6555 CrossRef CAS.
- Z. Gross and S. Nimri, J. Am. Chem. Soc., 1995, 117, 8021–8022 CrossRef CAS; J. T. Groves, Z. Gross and M. K. Stern, Inorg. Chem., 1994, 33, 5065–5072 CrossRef CAS; D. Ostovic and T. C. Bruice, J. Am. Chem. Soc., 1989, 111, 6511–6517 CrossRef.
- J. Bernadou, A.-S. Fabiano, A. Robert and B. Meunier, J. Am. Chem. Soc., 1994, 116, 9375–9376 CrossRef CAS.
- I. M. C. M. Rietjens, A. M. Osman, C. Veeger, O. Zakharieva, J. Antony, M. Grodzicki and A. X. Trautwein, J. Biol. Inorg. Chem., 1996, 1, 372–376 CrossRef CAS; A. M. Osman, J. Koerts, M. G. Boersma, S. Boeren, C. Veeger and I. M. C. M. Rietjens, Eur. J. Biochem., 1996, 240, 232–238 CrossRef CAS.
- A. W. Van der Made, W. Drenth and R. J. M. Nolte, Recl. Trav. Chim. Pays-Bas, 1987, 106, 330 Search PubMed; E. Guilmet and B. Meunier, Nouv. J. Chim., 1982, 6, 511–513 Search PubMed.
- P. Battioni, J. P. Renaud, J. F. Bartoli, M. Reina-Artiles, M. Fort and D. Mansuy, J. Am. Chem. Soc., 1988, 110, 8462–8470 CrossRef CAS.
- J. T. Groves and Y. Watanabe, J. Am. Chem. Soc., 1988, 110, 8443–8452 CrossRef CAS.
- E. Okochi and M. Mochikuzi, Chem. Pharm. Biol., 1995, 43, 2173–2176 Search PubMed.
- H. H. Ruf and P. J. Wende, J. Am. Chem. Soc., 1977, 99, 5499–5500 CrossRef CAS.
- L. Ricard, M. Schappacher, R. Weiss, R. Montiel-Montoya, E. Bill, U. Gonser and A. Trautwein, Nouv. J. Chim., 1983, 7, 405–408 Search PubMed.
- W.-D. Woggon, Top. Curr. Chem., 1996, 184, 39–96 Search PubMed; H. Patzelt and W.-D. Woggon, Helv. Chim. Acta, 1992, 75, 523–538 CrossRef CAS.
- T. Higuchi and M. Hirobe, J. Mol. Catal. A: Chem., 1996, 113, 403–422 CrossRef CAS; Y. Urano, T. Higuchi, M. Hirobe and T. Nagano, J. Am. Chem. Soc., 1997, 119, 12008–12009 CrossRef CAS.
- N. Ueyama, N. Nishikawa, Y. Yamada, T. Okamara and A. Nakamura, J. Am. Chem. Soc., 1996, 118, 12826–12827 CrossRef CAS; N. Ueyama, N. Nishikawa, Y. Yamada, T. Okamura, S. Oka, H. Sakurai and A. Nakamura, Inorg. Chem., 1998, 37, 2415–2421 CrossRef CAS.
- N. Suzuki, T. Higuchi, Y. Urano, K. Kikuchi, H. Uekusa, Y. Ohashi, T. Uchida, T. Kitagawa and T. Nagano, J. Am. Chem. Soc., 1999, 121, 11571–11572 CrossRef CAS.
- J. P. Collman, T. R. Halbert and K. S. Suslick, in Metal ions in Biology, ed. T. G. Spiro, Vol. 2, Search PubMed; Metal Ion Activation of Dioxygen, John Wiley and Sons, New York, 1980, pp. 1–72; Search PubMed; J. P. Collman and L. Fu, Acc. Chem. Res., 1999, 32, 455–463 Search PubMed.
- T. G. Traylor and P. S. Traylor, in Active Oxygen in Biochemistry, ed. J. S. Valentine, C. S. Foote, A. Greenberg and J. F. Liebman, Blackie Academic & Professional, London, 1995, pp. 84–187. Search PubMed.
- M. C. Feiters, in Comprehensive Supramolecular Chemistry, executive eds. J.-M. Lehn, J. L. Atwood, J. E. D. Davies, D. D. Macnicol and F. Vögtle, Vol. 10, Search PubMed; Supramolecular Technology and Applications, volume ed. D. N. Reinhoudt, 1996, pp. 267–360; Search PubMed; Z. Gross, L. Simkhovuch, G. N. Nitsa and I. Saltsman, Org. Lett., 1999, 1, 2077–2080 Search PubMed; Gloriana Reginato, L. Di Bari, P. Salvadori and R. Guilard, Eur. J. Org. Chem., 2000, 7, 1165–1171 Search PubMed; J. P. Collman, M. Zhong, Z. Wang, M. Rapta and E. Rose, Chem. Commun., 1999, 18, 1783–1784 CrossRef CAS.
- R. Breslow and S. D. Dong, Chem. Rev., 1998, 98, 1997–2011 CrossRef CAS; I. Tabushi, Coord. Chem. Rev., 1988, 86, 1–422 CrossRef CAS.
- R. Breslow, Proc. Natl. Acad. Sci. U.S.A., 1997, 94, 11156–11158 CrossRef CAS; R. Breslow, Y. Huang and X. Zhang, J. Am. Chem. Soc., 1997, 199, 4535–4536 CrossRef CAS.
- Y. Kuroda, T. Sera and H. Ogoshi, J. Am. Chem. Soc., 1991, 113, 2793–2794 CrossRef CAS.
- L. Weber, R. Hommel, J. Behling, G. Haufe and H. Hennig, J. Am. Chem. Soc., 1994, 116, 2400–2408 CrossRef CAS.
- D. R. Benson, R. Valentekovich and F. Diederich, Angew. Chem., Int. Ed. Engl., 1990, 29, 191–193 CrossRef; D. R. Benson, R. Valentekovich, S. W. Tam and F. Diederich, Helv. Chim. Acta, 1993, 76, 2034–2060 CrossRef CAS.
- L. Pauling, Nature, 1948, 161, 707–709 CrossRef CAS.
- X. Liu, S. Chen, Y. Feng, G. Gao and T. Yang, Ann. N. Y. Acad. Sci., 1998, 864, 273–275 Search PubMed; A. W. Schwabacher, M. I. Weinhouse, M.-T. M. Auditor and R. A. Lerner, J. Am. Chem. Soc., 1989, 111, 2344–2346 CrossRef CAS.
- A. G. Cochran and P. G. Schultz, Science, 1990, 249, 781–783 CAS.
- A. Harada, H. Fukushima, K. Shiotzuki, H. Yamaguchi, F. Oka and M. Kamachi, Inorg. Chem., 1997, 36, 6099–6102 CrossRef CAS.
- E. Keinan, E. Benory, S. C. Sinha, A. Sinha-Bagchi, D. Eren, Z. Eshhar and B. S. Green, Inorg. Chem., 1992, 31, 5433–5438 CrossRef CAS.
- S. Nimri and E. Keinan, J. Am. Chem. Soc., 1999, 121, 8978–8982 CrossRef CAS.
- J.-L. Reymond, G. K. Jahangiri, C. Stoudt and R. A. Lerner, J. Am. Chem. Soc., 1993, 115, 3909–3917 CrossRef CAS.
- A. Koch, J.-L. Reymond and R. A. Lerner, J. Am. Chem. Soc., 1994, 116, 803–804 CrossRef CAS.
- L. C. Hsieh, J. C. Stephans and P. G. Schultz, J. Am. Chem. Soc., 1994, 116, 2167–2168 CrossRef CAS.
- I. Tabushi and N. Koga, J. Am. Chem. Soc., 1979, 101, 6456–6458 CrossRef CAS; I. Tabushi and A. Yazaki, J. Am. Chem. Soc., 1981, 103, 7371–7373 CrossRef CAS.
- J. P. Hage, J. R. Schnelten and D. T. Sawyer, Bioorg. Med. Chem., 1996, 4, 821–824 CrossRef CAS; Y. Tsuda, K. Takahashi, T. Yamaguchi, S. Matsui, T. Komura and I. Nishiguchi, J. Mol. Catal. A: Chem., 1999, 138, 145–153 CrossRef CAS.
- A. B. Sorokin, A. M. Khenkin, S. A. Marakushev, A. E. Shilov and A. A. Shteinman, Dokl. Phys. Chem. (Engl. Trans.), 1984, 29, 1101–1102 Search PubMed.
- J. T. Groves and R. Neumann, J. Am. Chem. Soc., 1987, 109, 5045–5047 CrossRef CAS; J. T. Groves and R. Neumann, J. Am. Chem. Soc., 1989, 111, 2900–2909 CrossRef CAS.
- J. T. Groves and S. B. Ungashe, J. Am. Chem. Soc., 1990, 112, 7796–7797 CrossRef CAS; S. B. Ungashe and J. T. Groves, in Models in Inorganic Biochemistry, ed. G. L. Eichhorn and L. G. Marzilli, Prentice Hall, Englewood Cliffs, New Jersey, 1993, vol. 9, pp. 317–351. Search PubMed.
- J. Van Esch, M. F. M. Roks and R. J. M. Nolte, J. Am. Chem. Soc., 1986, 108, 6093–6094 CrossRef CAS.
- P. A. Gosling, M. A. M. Hoffmann, J. H. van Esch and R. J. M. Nolte, J. Chem. Soc., Chem. Commun., 1993, 472–473 RSC.
- A. P. H. J. Schenning, D. H. W. Hubert, J. H. van Esch, M. C. Feiters and R. J. M. Nolte, Angew. Chem., Int. Ed. Engl., 1994, 33, 2468–2470 CrossRef.
- A. P. H. J. Schenning, D. H. W. Hubert, M. C. Feiters and R. J. M. Nolte, Langmuir, 1996, 12, 1572–1577 CrossRef CAS.
- A. P. H. J. Schenning, J. H. Lutje Spelberg, D. H. W. Hubert, M. C. Feiters and R. J. M. Nolte, Chem. Eur. J., 1998, 4, 871–880 CrossRef CAS.
- (a) D. L. Jiang and T. Aida, Chem. Commun., 1996, 1523–1524 RSC; (b) T. Aida, J. Am. Chem. Soc., 1998, 120, 10895–10901 CrossRef; (c) J. P. Collman, L. Fu, A. Zingg and F. Diederich, Chem. Commun., 1997, 193–194 RSC.
- J. L. M. van Nunen, B. F. B. Folmer and R. J. M. Nolte, J. Am. Chem. Soc., 1997, 119, 1283–1291; J. L. M. van Nunen and R. J. M. Nolte, J. Chem. Soc., Perkin Trans. 1, 1997, 1473–1481 Search PubMed.
| This journal is © The Royal Society of Chemistry 2000 |