A spirooxazine showing crystalline state photochromism
Sophie
Bénard
and
Pei
Yu
*
Laboratoire de Chimie Inorganique, Bat. 420, UMR 8613, Université Paris Sud, F-91405, Orsay, France.. E-mail: yupei@icmo.u-psud.fr
First published on 6th January 2000
Abstract
A cationic spiroquinoxazine (SO2) is found to be photochromic both in solution and in the crystalline state.
Among the large number of investigated so far photochromic molecules,1,2 few have been found to be photochromic in the crystalline state.3–8 Photochromic organic crystals are interesting not only for the design of new materials for optical data processing and storage,4,6 but also because the photoinduced molecular transformations might be used to gain control over other physical properties in the solid state. For instance, the photoswitching of NLO properties has been recently achieved in a photochromic organic crystal.9 Spirooxazines (SO) are known to offer remarkable stability towards photo-fatigue in solution and in various matrices,10–14 but to the best of our knowledge none of them has been reported to be photochromic in the crystalline state. We report herein the first spirooxazine to exhibit photochromism both in solution and in the pure crystalline state.
N-Methylation of spiroquinoxazine10,12c (SO1) with MeI in THF yielded the yellow microcrystalline cationic SO15a (SO2, see Scheme 1).
 | ||
| Scheme 1 Scheme 1 | ||
Photochromism of spirooxazines is based on photocleavage of the C(spiro)–O bond, giving rise to an equilibrium between a colorless or weakly colored closed form and a strongly colored open form. The usually less stable open form reverts back to the closed form both photochemically and thermally. In the present case such an equilibrium is shown in Scheme 1.
Electronic absorption spectral changes of SO2 upon UV irradiation (365 nm)15b in CHCl3 are depicted in Fig. 1. The main characteristics of the closed and open forms as well as the thermal color fading rate (k) of the latter are reported in Table 1.
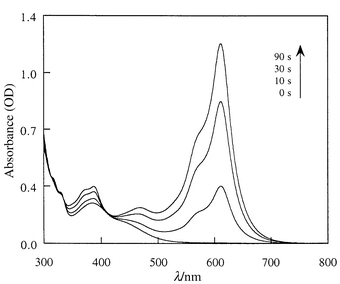 | ||
| Fig. 1 UV–VIS spectral changes of SO2 in CHCl3 solution (5 × 10−5M, room temperature) upon UV irradiation. | ||
The photochromic properties of SO2 are markedly different from those of SO1. First, at room temperature the thermal color decay of the open form of SO2, which follows a single exponential equation in the same manner as SO1, is very slow. Depending on the solvent, the constant k of this decay is about 103 to 104 times smaller than that of SO1. In other words, the N-methylation of the quinoline on the oxazine moiety stabilizes in a dramatic way the open form. Second, in contrast to the parent SO1 and other SO molecules, which are known to show positive solvatochromism,10,12a the open form of SO2 is characterized by a negative solvatochromism (Table 1), indicating that its ground state is more polar than the exited one. These two observations suggest a predominantly zwitterionic structure for the open form of SO2 rather than the less polar quinoidal one.
Irradiation of a microcrystalline sample of SO2 was carried out using an ultra-thin pressed pellet. The UV–VIS absorption spectra after different irradiation times are shown in Fig. 2. Upon UV irradiation (365 nm) a broad absorption band appeared between 550 and 650 nm and continuously increased while the initial yellow pellet turned green. When the sample was left in the dark at room temperature after irradiation, the green color faded slowly and the yellow colour returned very slowly. In contrast, visible light (550 nm) irradiation considerably accelerated the color decay. This behavior is similar to that observed in solution and indicates clearly that SO2 is photochromic in the crystalline state.
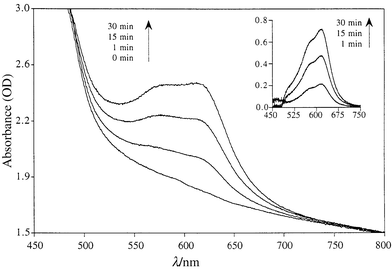 | ||
| Fig. 2 Crystalline state UV–VIS spectral changes of SO2 upon UV irradiation. Insert shows the differential optical density between the spectra before and after UV irradiation. | ||
By monitoring the decay of the absorption band in the visible region at room temperature, the thermal decoloration rate of the thin pellet was found to deviate significantly from first order kinetics, but can be fairly well fitted to a biexponential equation with k1 = 1.6 × 10−4 s−1 and k2 = 5.8 × 10−6 s−1. This behavior can be tentatively interpreted as the result of two different environments surrounding the open form of SO2 in the bulk sample. The open form molecules located near the surface of the solid are surrounded mostly by similar open form molecules. On the contrary an open form molecule lying deeper inside the bulk would have mainly closed form molecules as neighbors.
Finally, it is worth pointing out that the color change was not accompanied by any significant structure modifications, as the X-ray powder pattern of the irradiated sample did not show any detectable changes as compared to that of the initial one, while the large differences in geometry between the closed and open forms would lead one to expect rather large structure changes. A possible explanation would be that under current experimental conditions the photochemical process is mainly limited to the surface of the bulk sample, so the amount of the photoinduced open form is too small to give any significant changes in the X-ray powder pattern.
The reasons why SO2 shows crystalline state photochromism are not yet clearly understood. Nevertheless, the large stabilization of a predominantly zwitterionic open form brought about by the N-methylation of the quinoline ring seems to be an important factor in permitting solid state photochromism. On the other hand, the presence of iodide ions could result in more space or free volume in the crystal structure of SO2 as compared to the parent SO1. In other words, the anion, although not directly involved in the photochemical process, may have a kind of dilution or matrix-like effect on the cationic photoactive spirooxazine. These remarks are supported by the following observation: two derivatives of SO2, prepared by replacing the iodide anion in SO2 by nitrate or toluene-p-sulfonate, were also found to show similar behavior. This rather anion-independent behavior strongly suggests that crystalline state photochromism in this class of compounds depends mainly on the cationic nature of the photoactive species, and the role of the anion is probably minor. This particular feature would be of interest in the perspective of associating solid state photochromism with other properties that could be introduced through the anion.
Acknowledgements
We are grateful to Dr Keitaro Nakatani and Professor René Clément for many helpful discussions.References
-
Photochromism. Molecules and Systems, ed. H. Durr and H.
Bouas-Laurent, Elsevier, Amsterdam,
1990. Search PubMed
.
- B. L. Feringa, W. F. Jager and B. de Lange, Tetrahedron, 1993, 49, 8267 CrossRef CAS
- E. Hadjoudis, in ref. 1, ch. 17..
- M. Irie and K. Uchida, Bull. Chem. Soc. Jpn., 1998, 71, 985 CAS
-
S. Kobatake,
T. Yamada,
K. Uchida,
N. Kato and
M. Irie, J. Am.
Chem. Soc., 1999, 121,
2380 and references cited therein. Search PubMed
- Y. Eichen, J. M. Lehn, M. Scherl, D. Haarer, J. Fischer, A. DeCian, A. Corval and H. P. Trommsdorff, Angew. Chem., Int. Ed. Engl., 1995, 34, 2530 CrossRef CAS
- Y. H. Zhou, W. E. Baker, P. M. Kazmaier and E. Buncel, Can. J. Chem., 1998, 76, 884 CrossRef CAS
- K. Okada, K. Imamura, M. Oda, M. Kozaki, Y. Morimoto, K. Ishino and K. Tashiro, Chem. Lett., 1998, 891 CrossRef CAS
- K. Nakatani and J. A. Delaire, Chem. Mater., 1997, 9, 2682 CrossRef CAS
- N. Y. C. Chu, in ref. 1, ch. 10..
- S. Kawauchi, H. Yoshida, N. Yamashina, M. Ohira, S. Saeda and M. Irie, Bull. Chem. Soc. Jpn., 1990, 63, 267 CAS
-
(a) E. Pottier, R. Dubest, R. Guglielmetti, P. Tardieu, A. Kellmann, F. Tfibel, P. Levoir and J. Aubard, Helv. Chim. Acta, 1990, 73, 303 CrossRef CAS
- J. Biteau, F. Chaput and J. P. Boilot, J. Phys. Chem., 1996, 100, 9024 CrossRef CAS
- B. Schaudel, C. Guermeur, C. Sanchez, K. Nakatani and J. Delaire, J. Mater. Chem., 1997, 7, 61 RSC
-
(a)
(a) Selected data for SO2: (calc. for
C22H22IN3O: C, 56.06; H, 4.70; N, 8.91; O,
3.39. Found: C, 55.92; H, 4.77; N, 8.98; O, 3.34%);
δH(CDCl3) 1.34 (s, 3 H), 1.38 (s, 3 H), 2.75
(s, 3 H), 4.90 (s, 3 H), 6.61 (d, 1 H), 6.97 (t, 1 H), 7.10 (d, 1 H), 7.22
(t, 1 H), 7.68 (d, 1 H), 7.90 (s, 1 H), 8.10 (dd, 1 H), 8.19 (d, 1H), 9.60
(d, 1H), 10.18 (d, 1H).
| This journal is © The Royal Society of Chemistry 2000 |