The preparation of a series of nitrostilbene ester compounds using micro reactor technology
Victoria Skeltona, Gillian M. Greenwaya, Stephen J. Haswell*a, Peter Styringa, David O. Morganb, Brian Warringtonc and Stephanie Y. F. Wongc
aDepartment of Chemistry, Faculty of Science and the Environment, University of Hull, Cottingham Road, Hull, UK HU6 7RX
bSmithKline Beecham Pharmaceuticals, Old Powder Mills, Tonbridge, Nr Leigh, Kent, UK TN11 9AN
cSmithKline Beecham Pharmaceuticals, New Frontiers Science Park (North), Third Avenue, Harlow, Essex, UK CM19 5AW
First published on 27th November 2000
Abstract
The synthesis of stilbene esters using Wittig chemistry has been used to illustrate the generic diversity micro reactors offer in terms of chemical control and rapid method development. The micro reactor consisted of a ‘T’ design based on channel geometries 200 μm wide and 100 μm deep, etched into borosilicate glass and sealed with a borosilicate top plate using a thermal bonding technique. The movement of the reagent and products was achieved using electroosmotic flow (EOF), assisted by the incorporation of micro porous silica frits within the micro-channels to allow accurate solution control. To optimise the operating conditions methyl 4-formylbenzoate, premixed with sodium methoxide, was reacted with 2-nitrobenzyl-triphenylphosphonium bromide in dry degassed MeOH using flow conditions for both reagents of 0.40 μL min−1 for 20 min. A product yield of 70% (2∶1 reaction stoichiometry with the aldehyde in excess) was obtained representing a 10% increase compared with the traditional batch synthesis. To demonstrate the capability of micro reactors to perform atom efficient synthesis a series of experiments based on an injection methodology (optimised to 30 s) were performed in the micro reactor at 1∶1 stoichiometry resulting in a yield of 59%. Finally, the capability of micro reactors to perform a series of analogue reactions was investigated. The yields for a further three aldehydes indicated that the technology will be suitable for the development of automated device to support the generation of combinatorial libraries and rapid high throughput synthetic methods.
Introduction
In recent years, interest in developing total analytical system (μ-TAS) has grown considerably1–10 as the reality of achieving a faster and cheaper means of performing chemical analysis has been demonstrated. Interestingly, the principles demonstrated by μ-TAS can readily be exploited to develop reactors at the micron scale for the synthesis of organic compounds. Whilst such devices offer a rapid and practically attractive approach to synthesis, the real potential lies in the possibility of achieving novel reaction control and product formation.11A strong research base is now developing in the field of micro reactor technology with a particular emphasis on gas phase catalytic reactions,12,13 and a growing interest in solution based chemistry.14 This trend in micro reactor methodology is reflected in the growing number of papers appearing in associated conferences.15 Micro reactor fabrication can be achieved using a variety of mediums, for example glass, metals and silicon, however, glass substrates with typical cross sections between 50 and 300 μm have been preferred for organic synthesis due to the chemical inertness, temperature stability, optical transparency and the ability to support electroosmotic flow (EOF).16
For the purpose of this study, the evaluation of a glass based micro reactor for synthetic chemistry has been investigated using as an example, the Wittig chemistry commonly used for vitamin A synthesis.17 The reaction (Fig. 1) allows the formation of a carbon to carbon bond by the reaction of a phosphorane or phosphonium ylide and an aldehyde or ketone to form an alkene and phosphine oxide. The Wittig reaction was investigated on a micro reactor with two specific aims, firstly to establish the criteria for achieving a solution based synthetic application and secondly, to investigate the potential for the technology to perform analogue-type chemistry. In this present case, the Wittig synthesis has been selected for its homogeneous solution properties, which produces a coloured intermediate (ylide) enabling solution profiling and diffusive mixing visualisation to be achieved, so gaining a greater understanding of system flow and reaction control.
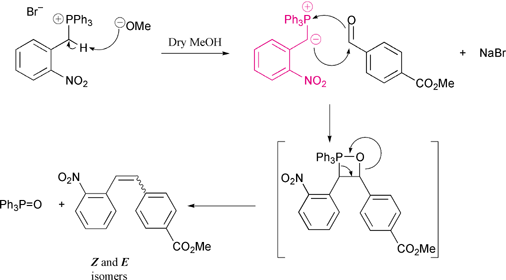 | ||
| Fig. 1 Reaction scheme for the coupling of 2-nitrobenzyltriphenylphosphonium bromide and methyl 4-formylbenzoate in a micro reactor under EOF. | ||
Experimental
All reagents were of analytical grade, unless otherwise stated and were used without further purification. The micro reactor was fabricated using photolithographic techniques (200 μm wide and 100 μm deep) and the microchannels included microporous silica frits18 positioned as shown in Fig. 2. The channels were sealed by annealing the patterned base plate to a 17 mm thick top plate (680 °C) using a microwave furnace (CEM microwave ashing system 300). The top plate included 3 mm internal diameter pre-drilled holes aligned at the ends of each channel to act as reservoirs and electrode supports. The final outer dimensions of the micro reactor were 20 mm × 20 mm square and 25 mm in depth. In addition, the total length of the channels was 20 mm from reservoir A to C.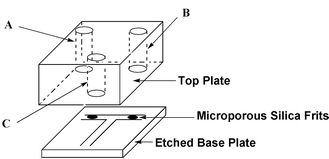 | ||
| Fig. 2 Schematic of the T-shaped manifold used in the reactor for the Wittig synthesis. | ||
The synthetic method was adapted from that previously reported by Hughes et al.19–20 In this paper, 2-nitrobenzyltriphenylphosphonium bromide was used as it produced a coloured intermediate (ylide), due to the presence of the nitro functional group. Visual observation of the ylide enabled the reaction and the fluidics of the system to be monitored via a microscope and CCD camera. The yield was also determined using high performance liquid chromatography (Zorbax C18 3.5 μm, 75 × 4.5 mm id, mobile phase composition: 0.1% trifluroacetic acid in water and 0.1% trifluroacetic acid in acetonitrile, using a gradient system 90% aqueous to 10% aqueous over 6 min with a flow rate of 3 ml min−1 at 40 °C).
Wittig synthesis based on 2∶1 stoichiometry
Prior to synthesis, the microchannels were primed with dry methanol (MeOH) removing any excess moisture from the channels and the microporous silica frits to reduce a hydrolysis side reaction. A standard solution of 2-nitrobenzyltriphenylphosphonium bromide (50 μL, 0.01 M) in dry MeOH was added into reservoir A of the micro reactor. Methyl 4-formylbenzoate (50 μL, 0.02 M, 2 eq.) was premixed with sodium methoxide (0.015 M) and 50 μL of the premixed solution was introduced into reservoir B. Dry MeOH (40 μL, dry, degassed) was introduced into reservoir C. Gold electrodes were placed in each reservoir (A + B positive, C ground) and an external voltage of 100–700 V was applied to channels A (+) and B (+) relative to reservoir C (−), see Fig. 2. This induced the continuous flow of methyl 4-formylbenzoate from B (+) to C (−) and 2-nitrobenzyltriphenylphosphonium bromide from A (+) to C. The total volume of the solutions in reservoir A and C at the end of each reaction was recorded and samples were taken for HPLC analysis. The flow rates over the voltage region 100 V to 700 V were determined using a volumetric approach with time. The yield obtained in the micro reactor was calculated from a knowledge of the input weight of 2-nitrobenzyltriphenylphosphonium bromide, the volume of solution in reservoirs A and C, before and after the reaction, measurement of the concentration of 2-nitrobenzyltriphenylphosphonium bromide in A and C at the end of the reaction and the concentration of the product in reservoir C.Using the optimised conditions obtained, three additional aldehydes with various functional diversities (supplied by SmithKline Beecham Pharmaceuticals, Harlow) were used to demonstrate the potential of micro reactors to perform a variety of reactions. The aldehydes chosen were 3-benzyloxybenzaldehyde, 2-naphthaldehyde and 5-nitrothiophene-2-carboxaldehyde. Each aldehyde was reacted using the optimum conditions established for the aldehyde methyl 4-formylbenzoate. For comparison purposes, each aldehyde was reacted in a batch mode using the method outlined below.
Batch scale methodology
Batch synthesis was also performed at 2∶1 and 1∶1 stoichiometry in order to make a comparison of the yields obtained with the micro reactor versus traditional synthetic methods. This was achieved by stirring 2-nitrobenzyltriphenylphosphonium bromide (0.01 M in MeOH) with sodium methoxide (0.015 M in dry degassed methanol) and each aldehyde in turn at various stoichiometric ratios (concentrations 0.02 M and 0.01 M) at room temperature for 20 min. The reaction parameters set for the micro reactor and the batch system allows the comparison of the reaction length (20 min), concentration, and the solvent of choice. The solution was then evaporated to dryness, purified by Prep HPLC and the yield (g) recorded.Wittig reaction based on 1∶1 stoichiometry
Traditional batch syntheses are typically performed with an excess of reagent, in this particular case the aldehyde is in excess relative to the phosphonium bromide. To establish the operating features of the micro reactor, for the reduced reaction stoichiometry of 1∶1, an injection profiling technique was adopted. This required the methyl 4-formylbenzoate solution to be continuously driven by EOF through the micro reactor from B to C whilst sample plugs of 2-nitrobenzyltriphenylphosphonium bromide were injected every min for a range of injection times (20 to 50 s) from channel A to C. Injection was initiated by the periodic application of an optimised voltage between A (+) and C (−) which injected slugs of the reagent (flow rate of 0.4 μL min−1 determined using volumetric flow rate with time). Repeat injections (20) for each injection time were carried out to generate sufficient product for off-chip analysis.Results and discussion
The 2∶1 stoichiometric study initially centred on evaluating the effect of reagent flow rates on the spatial location of the reaction intermediate by adjusting the voltage applied in reservoirs A and B relative to C. Whilst the initial data gave extremely high product yields several problems were experienced with the reaction. At high voltage (500–700 V) the purple colour observed from the ylide intermediate could clearly be seen being produced in reservoir C and the yields (approximately 50%) suggest the reaction was occurring in the reservoir rather than in the microchannels. Under such a regime, one of the fundamental benefits of the micro reactor, i.e., the spatial control, was not being exploited. The yield however was comparable with that obtained for the traditional batch methodology. As the voltage (flow rates) was decreased to less than 400 V, the yield increased dramatically, on average from 30 to 70%. The variation in the RSD of the yield at 100 V was however greater than 5% associated error due to the irreproducible generation of the flow rates for both reagents. However, at 400 V an optimum pumping voltage (this equated to a flow rate of 0.4 μL min−1) was found which offered well-controlled flow conditions in which the reaction was contained within the channels of the micro reactor.Fig. 3 summarises the results obtained for the micro reactor compared with the traditional batch process. In this case, the optimised micro reactor conditions were based on methyl 4-formybenzoate (A), which gave an overall product yield of 70% compared to 60% obtained for the batch method performed in this study.
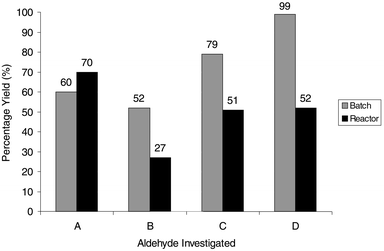 | ||
| Fig. 3 Summary of the product yields obtained for both micro reactor and traditional batch methods for four aldehydes based on the optimisation with methyl 4-formylbenzoate using 2∶1 stoichiometry. The aldehydes shown are methyl 4-formylbenzoate (A), 3-benzyloxybenzaldehyde (B), 2-naphthaldehyde (C) and 5-nitrothiophene-2-carboxaldehyde (D). | ||
Using the optimum parameters established for methyl 4-formylbenzoate, Fig. 3 summarises the remaining results obtained for three additional aldehydes. For all the aldehydes tested the yield obtained in the micro reactor (2∶1) were approximately half of those obtained by the traditional batch methodology. It is postulated that the reduction in yield may be due to the individual variation in flow dynamics through the micro reactor associated with each aldehyde, as described below.
Fig. 4 shows a series of photographs captured using an optical microscope. The images clearly indicate the diffusive flow profile of the reaction intermediate (ylide). Plate 1 shows the movement of reagents from the left (2-nitrobenzyltriphenylphosphonium bromide) and right hand (aldehyde and base) channels via EOF at 350 V. A clear diffusive reaction interface can be seen between the two reagents. However, due to poor flow control the formation of the ylide can be seen to be primarily occurring in the left-hand channel. This may be due to the zeta potential in the central channel being smaller than in the two feeder channels thus causing feedback of the reagent from B to A.21,22 In addition, the base, which is premixed with the aldehyde, has greater electrophoretic mobility compared to the aldehyde, see Plate 1 for ylide formation in the left hand channel. By slightly increasing the voltage and hence the flow rate in the left hand channel, the ‘over flow’ from the right hand channel can be corrected, see Plate 2. In this case the reaction can be spatially contained in the middle channel of the T reactor, see Fig. 2. Further adjustment of the voltages applied to the feed reservoirs allows complete diffusive mixing across the channel to be achieved, see Plate 3, demonstrating the EOF control possible in micro reactors. This small variation in reagent flow can significantly alter the position and the reaction conditions of the reagents in the channels. Thus when using a number of reagents, it is probable that each reagent will have a slightly different flow profile causing a disruption to the diffusive reaction interface. Such a situation will result in the reaction interface occurring in a non-optimum position for example, in either of the feed channels, where reaction conditions may not be ideal, resulting in reduced reaction yields. The results indicate that by in situ monitoring of the reaction and voltage feedback, an automated system could be developed enabling rapid method development and optimisation with high throughput synthetic capabilities.
 | ||
| Fig. 4 Photographs obtained from an optical microscope showing the Wittig reaction ylide intermediate formation under variable flow control. | ||
The 1∶1 stoichiometric study was carried out to investigate the effects on the overall solution yield of periodically injecting a known volume of 2-nitrobenzyltriphenylphosphoniuim bromide into a continuous stream of aldehyde, e.g. methyl 4-formylbenzoate. Previously, Fig. 3 illustrated a decrease in the yields for three of the aldehydes investigated in the micro reactor when compared to the traditional bulk reaction. By developing an injection profile (i.e. the optimisation of the injection time and interval) for the reaction, the surface area of the reaction is increased automatically which in theory should enhance the overall yield.13 In addition to increasing the overall yield, the fundamental features of the micro reactor, i.e., spatial and temporal control needed to be investigated. This was easily achievable by reducing the stoichiometry of the reaction, which in turn enabled the micro reactor to be evaluated for reagent efficient chemistry.
Fig. 5 summarises the yields obtained for the traditional batch reaction and micro reactor scale based on a 1∶1 stoichiometry. For the micro reactor the maximum yield of 59% was achieved using a 30 s injection length (i.e. one 30 s injection per min over a total of 20 min) from channel A into a continuous flow from B to C, see Fig. 2. Whilst there is a 10% reduction in the overall yield compared with the 2∶1 reaction, the micro reactor under the injection optimised conditions still produced a high product yield at the chemically more favourable 1∶1 stoichiometry. The 1∶1 stoichiometry results in a lower reagent consumption and reduced work up and sample purification.
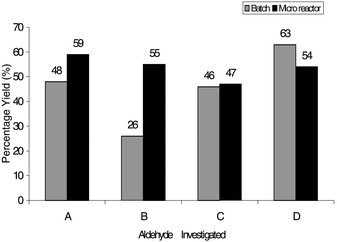 | ||
| Fig. 5 Summary of the product yields obtained for both micro reactor and traditional batch methods for four aldehydes based on the optimisation with methyl 4-formylbenzoate using 1∶1 stoichiometry. The aldehydes shown are methyl 4-formylbenzoate (A), 3-benzyloxybenzaldehyde (B), 2-naphthaldehyde (C) and 5-nitrothiophene-2-carboxaldehyde (D). | ||
Using the optimum reaction (reservoirs A and B at 400 V with an injection length of 30 s from reservoir A) conditions based on methyl 4-formylbenzoate, once again the additional three test aldehydes were reacted. For the reaction with aldehyde B (3-benzyloxybenzaldehyde) in the micro reactor, a 29% increase in reaction efficiency was obtained compared with the batch reaction performed using the same concentrations, solvent and reaction time. For the reaction using aldehyde C (2-naphthaldehyde), the micro reactor yield was comparable with the traditional batch methodology. Aldehyde D (5-nitrothiophene-2-carboxaldehyde) gave a 9% decrease in reaction efficiency, which was an improvement over the 2:1 data (47% reduction).
The micro reactor device allows the synthesis of nitro stilbene esters, which are comparable with the reactions obtained in a more traditional fashion. With respect to the variation in yields for the remaining aldehydes tested, it is worth stressing that the micro reactors generated products rapidly and in sufficient quantities to determine the products of each reaction. This attribute of micro reactors clearly has important implications in establishing rapid analogue based high throughput chemistries and so representing a significant approach for future developments in the field.
Conclusions
The micro reactor has demonstrated the synthetic novelty to achieve the rapid synthesis of nitro stilbene esters, and hence the potential generic diversity using simple homogeneous solution chemistry. The optimised micro reactor based on methyl 4-formylbenzoate on average gave a 10% increase in reaction efficiency for both 2∶1 and 1∶1 stoichiometry. In addition, the reactor could also be used as a tool for rapid reaction development and optimisation based on analogue chemistry.Due to the diverse reaction features of the micro reactor, a combinatorial screening device is currently being investigated illustrating the potential to synthesise a combinatorial library and on-chip screening. If the micro reactor device could be coupled with a separation chip, the Z and E isomers could be purified from the solution making the device suitable for synthetic development.
Acknowledgements
We thank the EPSRC and SmithKline Beecham Pharmaceuticals for financial support to VS in the form of a CASE studentship.References
- S. Cowen, Chem. Ind. (London), 1999, 15, 584 Search PubMed.
- T. McCreedy, Chem. Ind. (London), 1999, 15, 588 Search PubMed.
- D. Barrow, J. Cefai and S. Taylor, Chem. Ind. (London), 1999, 15, 591 Search PubMed.
- S. J. Haswell, Analyst, 1997, 122, 1R RSC.
- Proceedings of the micro total analytical systems ‘98 workshop, ed. D. J. Harrison and A. van den Berg, Kluwer Academic Publishers, Drodrecht, 1998. Search PubMed.
- A. Manz, D. J. Harrison, E. Verpoorte, J. C. Fettinger, H. Ludi and H. M. Widmer, Chimia, 1991, 45, 103 Search PubMed.
- A. Manz, D. J. Harrison, E. Verpoorte and H. M. Widmer, Adv. Chromarogr, 1993, 33, 1 Search PubMed.
- A. Manz, C. S. Effenhauser, N. Barggraf, E. Verpoorte, D. E. Raymond and H. M. Widmer, Analysis Mag, 1994, 22, M25 Search PubMed.
- D. J. Harrison, K. Fluri, K. Seiler, Z. H. Fan, C. S. Effenhauser and A. Manz, Science, 1993, 261, 895 CrossRef CAS.
- S. C. Jacobson, R. Hergenroder, L. B. Koutny and J. M. Ramsey, Anal. Chem., 1994, 66, 1114 CrossRef CAS.
- P. D. I. Fletcher and S. J. Haswell, Chem. Br., 1999, 38 Search PubMed.
- W. Ehrfeld and H. Lehr, Radiat. Phys. Chem., 1995, 45, 349 CrossRef CAS.
- R. D. Chambers and R. C. H. Spink, Chem. Commun., 1999, 10, 883 RSC.
- G. M. Greenway, S. J. Haswell, D. O. Morgan, V. Skelton and P. Styring, Sens. Actuators B, 2000, 63, 153 CrossRef.
- IMRET 4: 4th International Conference on Microreaction Technology, Topical conference proceedings, March 5–9 2000, Atlanta, GA..
- P. D. I. Fletcher, S. J. Haswell and V. N. Paunov, Analyst, 1999, 124, 1273 RSC.
- J. McMurray, Organic Chemistry, 3rd edn., 1992, Wadsworth Inc..
- P. D. Christensen, S. W. P. Johnson, T. McCreedy, V. Skelton and N. G. Wilson, Anal. Commun., 1998, 35, 341 RSC.
- I. Hughes, W. P. Nolan and R. A. Raphael, J. Chem. Soc. Perkin Trans., 1990, 1, 2475 Search PubMed.
- I. Hughes, Tett. Lett., 1996, 37(42), 7595 CrossRef CAS.
- R. S. Ramsey and J. M. Ramsey, Anal. Chem., 1997, 69, 1174 CrossRef CAS.
- L. Hu, D. J. Harrison and J. H. Masliyah, Colloid Interface Sci., 1999, 215, 300 Search PubMed.
| This journal is © The Royal Society of Chemistry 2001 |