Introduction to multivariate calibration in analytical chemistry†
Richard G. Brereton
School of Chemistry, University of Bristol, Cantock’s Close, Bristol, UK BS8 1TS
First published on 31st October 2000
 Richard G. Brereton Richard G. Brereton | Richard Brereton performed his undergraduate, postgraduate and postdoctoral studies in the University of Cambridge, and moved to Bristol in 1983, where he is now a Reader. He has published 169 articles, 85 of which are refereed papers, and his work has been cited over 1100 times. He has presented over 50 public invited lectures. He is currently chemometrics columnist for the webzine the Alchemist. He is author of one text, and editor of three others. His interests encompass multivariate curve resolution, calibration, experimental design and pattern recognition, primarily in the area of coupled chromatography, as applied to a wide variety of problems including pharmaceutical impurity monitoring, rapid reaction kinetics, food and biological chemistry. |
1 Introduction
1.1 Overview
Multivariate calibration has historically been a major cornerstone of chemometrics as applied to analytical chemistry. However, there are a large number of diverse schools of thought. To some, most of chemometrics involves multivariate calibration. Certain Scandinavian and North American groups have based much of their development over the past two decades primarily on applications of the partial least squares (PLS) algorithm. At the same time, the classic text by Massart and co-workers1 does not mention PLS, and multivariate calibration is viewed by some only as one of a large battery of approaches to the interpretation of analytical data. In Scandinavia, many use PLS for almost all regression problems (whether appropriate or otherwise) whereas related methods such as multiple linear regression (MLR) are more widely used by mainstream statisticians.There has developed a mystique surrounding PLS, a technique with its own terminology, conferences and establishment. Although originally developed within the area of economics, most of its prominent proponents are chemists. There are a number of commercial packages on the market-place that perform PLS calibration and result in a variety of diagnostic statistics. It is, though, important to understand that a major historic (and economic) driving force was near infrared spectroscopy (NIR), primarily in the food industry and in process analytical chemistry. Each type of spectroscopy and chromatography has its own features and problems, so much software was developed to tackle specific situations which may not necessarily be very applicable to other techniques such as chromatography or NMR or MS. In many statistical circles NIR and chemometrics are almost inseparably intertwined. However, other more modern techniques are emerging even in process analysis, so it is not at all certain that the heavy investment on the use of PLS in NIR will be so beneficial in the future. Despite this, chemometric approaches to calibration have very wide potential applicability throughout all areas of quantitative analytical chemistry.
There are very many circumstances in which multivariate calibration methods are appropriate. The difficulty is that to develop a very robust set of data analytical techniques for a particular situation takes a large investment in resources and time, so the applications of multivariate calibration in some areas of science are much less well established than in others. It is important to distinguish the methodology that has built up around a small number of spectroscopic methods such as NIR, from the general principles applicable throughout analytical chemistry. This article will concentrate on the latter. There are probably several hundred favourite diagnostics available to the professional user of PLS e.g. in NIR spectroscopy, yet each one has been developed with a specific technique or problem in mind, and are not necessarily generally applicable to all calibration problems. The untrained user may become confused by these statistics; indeed he or she may have access to only one specific piece of software and assume that the methods incorporated into that package are fairly general or well known, and may even inappropriately apply diagnostics that are not relevant to a particular application.
There are a whole series of problems in analytical chemistry for which multivariate calibration is appropriate, but each is very different in nature.
1. The simplest is calibration of the concentration of a single compound using a spectroscopic or chromatographic method, an example being determining the concentration of chlorophyll by EAS (electronic absorption spectroscopy).2 Instead of using one wavelength (as is conventional for the determination of molar absorptivity or extinction coefficients), multivariate calibration involves using all or several of the wavelengths.
2. A more complex situation is a multi-component mixture where all pure standards are available, such as a mixture of four pharmaceuticals.3 It is possible to control the concentration of the reference compounds, so that a number of carefully designed mixtures can be produced in the laboratory. Sometimes the aim is to see whether a spectrum of a mixture can be employed to determine individual concentrations, and, if so, how reliably. The aim may be to replace a slow and expensive chromatographic method by a rapid spectroscopic approach. Another rather different aim might be impurity monitoring,4 how well the concentration of a small impurity may be determined, for example, buried within a large chromatographic peak.
3. A different approach is required if only the concentration of a portion of the components is known in a mixture, for example, the polyaromatic hydrocarbons within coal tar pitch volatiles.5 In the natural samples there may be tens or hundreds of unknowns, but only a few can be quantified and calibrated. The unknown interferents cannot necessarily be determined and it is not possible to design a set of samples in the laboratory containing all the potential components in real samples. Multivariate calibration is effective provided that the range of samples used to develop the model is sufficiently representative of all future samples in the field. If it is not, the predictions from multivariate calibration could be dangerously inaccurate. In order to protect against samples not belonging to the original dataset, a number of approaches for determination of outliers have been developed.
4. A final case is where the aim of calibration is not so much to determine the concentration of a particular compound but a group of compounds, for example protein in wheat.6 The criteria here become fairly statistical and the methods will only work if a sufficiently large and adequate set of samples are available. However, in food chemistry if the supplier of a product comes from a known source that is unlikely to change, it is often adequate to set up a calibration model on this training set.
There are many pitfalls in the use of calibration models, perhaps the most serious being variability in instrument performance over time. Each instrument has different characteristics and on each day and even hour the response can vary. How serious this is for the stability of the calibration model needs to be assessed before investing a large effort. Sometimes it is necessary to reform the calibration model on a regular basis, by running a standard set of samples, possibly on a daily or weekly basis. In other cases multivariate calibration gives only a rough prediction, but if the quality of a product or the concentration of a pollutant appears to exceed a certain limit, then other more detailed approaches can be used to investigate the sample. For example, on-line calibration in NIR can be used for screening a manufactured sample, and any dubious batches investigated in more detail using chromatography.
There are many excellent articles and books on multivariate calibration which provide greater details about the algorithms.7–14 This article will compare the basic methods, illustrated by case studies, and will also discuss more recent developments such as multiway calibration and experimental design of the training set. There are numerous software packages available, including Piroutte,15 Unscrambler,16 SIMCA17 and Matlab Toolkit18 depending on the user’s experience. However, many of these packages contain a large number of statistics that may not necessarily be relevant to a particular problem, and sometimes force the user into a particular mode of thought. For the more computer based chemometricians, using Matlab for developing applications allows a greater degree of flexibility. It is important to recognise that the basic algorithms for multivariate calibration are, in fact, extremely simple, and can easily be implemented in most environments, such as Excel, Visual Basic or C.
1.2 Case study 1
The first and main case study for this application is of the electronic absorption spectra (EAS) of ten polyaromatic hydrocarbons (PAHs). Table 1 is of the concentrations of these PAHs in 25 spectra (dataset A) recorded at 1 nm intervals between 220 and 350 nm, forming a matrix which is often presented as having 25 rows (individual spectra) and 131 columns (individual wavelengths). The spectra are available as Electronic Supplementary Information (ESI Table S1†). The aim is to determine the concentration of an individual PAH in the mixture spectra.| Polyarene conc./mg L−1 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Spectrum | Py | Ace | Anth | Acy | Chry | Benz | Fluora | Fluore | Nap | Phen |
| a Abbreviations for PAHs: Py = pyrene; Ace = acenaphthene; Anth = anthracene; Acy = acenaphthylene; Chry = chrysene; Benz = benzanthracene; Fluora = fluoranthene; Fluore = fluorene; Nap = naphthalene; Phen = phenanthrene. | ||||||||||
| 1 | 0.456 | 0.120 | 0.168 | 0.120 | 0.336 | 1.620 | 0.120 | 0.600 | 0.120 | 0.564 |
| 2 | 0.456 | 0.040 | 0.280 | 0.200 | 0.448 | 2.700 | 0.120 | 0.400 | 0.160 | 0.752 |
| 3 | 0.152 | 0.200 | 0.280 | 0.160 | 0.560 | 1.620 | 0.080 | 0.800 | 0.160 | 0.118 |
| 4 | 0.760 | 0.200 | 0.224 | 0.200 | 0.336 | 1.080 | 0.160 | 0.800 | 0.040 | 0.752 |
| 5 | 0.760 | 0.160 | 0.280 | 0.120 | 0.224 | 2.160 | 0.160 | 0.200 | 0.160 | 0.564 |
| 6 | 0.608 | 0.200 | 0.168 | 0.080 | 0.448 | 2.160 | 0.040 | 0.800 | 0.120 | 0.940 |
| 7 | 0.760 | 0.120 | 0.112 | 0.160 | 0.448 | 0.540 | 0.160 | 0.600 | 0.200 | 0.118 |
| 8 | 0.456 | 0.080 | 0.224 | 0.160 | 0.112 | 2.160 | 0.120 | 1.000 | 0.040 | 0.118 |
| 9 | 0.304 | 0.160 | 0.224 | 0.040 | 0.448 | 1.620 | 0.200 | 0.200 | 0.040 | 0.376 |
| 10 | 0.608 | 0.160 | 0.056 | 0.160 | 0.336 | 2.700 | 0.040 | 0.200 | 0.080 | 0.118 |
| 11 | 0.608 | 0.040 | 0.224 | 0.120 | 0.560 | 0.540 | 0.040 | 0.400 | 0.040 | 0.564 |
| 12 | 0.152 | 0.160 | 0.168 | 0.200 | 0.112 | 0.540 | 0.080 | 0.200 | 0.120 | 0.752 |
| 13 | 0.608 | 0.120 | 0.280 | 0.040 | 0.112 | 1.080 | 0.040 | 0.600 | 0.160 | 0.376 |
| 14 | 0.456 | 0.200 | 0.056 | 0.040 | 0.224 | 0.540 | 0.120 | 0.800 | 0.080 | 0.376 |
| 15 | 0.760 | 0.040 | 0.056 | 0.080 | 0.112 | 1.620 | 0.160 | 0.400 | 0.080 | 0.940 |
| 16 | 0.152 | 0.040 | 0.112 | 0.040 | 0.336 | 2.160 | 0.080 | 0.400 | 0.200 | 0.376 |
| 17 | 0.152 | 0.080 | 0.056 | 0.120 | 0.448 | 1.080 | 0.080 | 1.000 | 0.080 | 0.564 |
| 18 | 0.304 | 0.040 | 0.168 | 0.160 | 0.224 | 1.080 | 0.200 | 0.400 | 0.120 | 0.118 |
| 19 | 0.152 | 0.120 | 0.224 | 0.080 | 0.224 | 2.700 | 0.080 | 0.600 | 0.040 | 0.940 |
| 20 | 0.456 | 0.160 | 0.112 | 0.080 | 0.560 | 1.080 | 0.120 | 0.200 | 0.200 | 0.940 |
| 21 | 0.608 | 0.080 | 0.112 | 0.200 | 0.224 | 1.620 | 0.040 | 1.000 | 0.200 | 0.752 |
| 22 | 0.304 | 0.080 | 0.280 | 0.080 | 0.336 | 0.540 | 0.200 | 1.000 | 0.160 | 0.940 |
| 23 | 0.304 | 0.200 | 0.112 | 0.120 | 0.112 | 2.700 | 0.200 | 0.800 | 0.200 | 0.564 |
| 24 | 0.760 | 0.080 | 0.168 | 0.040 | 0.560 | 2.700 | 0.160 | 1.000 | 0.120 | 0.376 |
| 25 | 0.304 | 0.120 | 0.056 | 0.200 | 0.560 | 2.160 | 0.200 | 0.600 | 0.080 | 0.752 |
A second dataset consisting of another 25 spectra, whose concentrations are given in Table 2, will also be employed where necessary (dataset B). The full data are available as Electronic Supplementary Information (ESI Table S2†). Most calibration will be performed on dataset A.
| Polyarene conc./mg L−1 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Spectrum | Py | Ace | Anth | Acy | Chry | Benz | Fluora | Fluore | Nap | Phen |
| 1 | 0.456 | 0.120 | 0.168 | 0.120 | 0.336 | 1.620 | 0.120 | 0.600 | 0.120 | 0.564 |
| 2 | 0.456 | 0.040 | 0.224 | 0.160 | 0.560 | 2.160 | 0.120 | 1.000 | 0.040 | 0.188 |
| 3 | 0.152 | 0.160 | 0.224 | 0.200 | 0.448 | 1.620 | 0.200 | 0.200 | 0.040 | 0.376 |
| 4 | 0.608 | 0.160 | 0.280 | 0.160 | 0.336 | 2.700 | 0.040 | 0.200 | 0.080 | 0.188 |
| 5 | 0.608 | 0.200 | 0.224 | 0.120 | 0.560 | 0.540 | 0.040 | 0.400 | 0.040 | 0.564 |
| 6 | 0.760 | 0.160 | 0.168 | 0.200 | 0.112 | 0.540 | 0.080 | 0.200 | 0.120 | 0.376 |
| 7 | 0.608 | 0.120 | 0.280 | 0.040 | 0.112 | 1.080 | 0.040 | 0.600 | 0.080 | 0.940 |
| 8 | 0.456 | 0.200 | 0.056 | 0.040 | 0.224 | 0.540 | 0.120 | 0.400 | 0.200 | 0.940 |
| 9 | 0.760 | 0.040 | 0.056 | 0.080 | 0.112 | 1.620 | 0.080 | 1.000 | 0.200 | 0.752 |
| 10 | 0.152 | 0.040 | 0.112 | 0.040 | 0.336 | 1.080 | 0.200 | 1.000 | 0.160 | 0.940 |
| 11 | 0.152 | 0.080 | 0.056 | 0.120 | 0.224 | 2.700 | 0.200 | 0.800 | 0.200 | 0.564 |
| 12 | 0.304 | 0.040 | 0.168 | 0.080 | 0.560 | 2.700 | 0.160 | 1.000 | 0.120 | 0.752 |
| 13 | 0.152 | 0.120 | 0.112 | 0.200 | 0.560 | 2.160 | 0.200 | 0.600 | 0.160 | 0.376 |
| 14 | 0.456 | 0.080 | 0.280 | 0.200 | 0.448 | 2.700 | 0.120 | 0.800 | 0.080 | 0.376 |
| 15 | 0.304 | 0.200 | 0.280 | 0.160 | 0.560 | 1.620 | 0.160 | 0.400 | 0.080 | 0.188 |
| 16 | 0.760 | 0.200 | 0.224 | 0.200 | 0.336 | 2.160 | 0.080 | 0.400 | 0.040 | 0.376 |
| 17 | 0.760 | 0.160 | 0.280 | 0.120 | 0.448 | 1.080 | 0.080 | 0.200 | 0.080 | 0.564 |
| 18 | 0.608 | 0.200 | 0.168 | 0.160 | 0.224 | 1.080 | 0.040 | 0.400 | 0.120 | 0.188 |
| 19 | 0.760 | 0.120 | 0.224 | 0.080 | 0.224 | 0.540 | 0.080 | 0.600 | 0.040 | 0.752 |
| 20 | 0.456 | 0.160 | 0.112 | 0.080 | 0.112 | 1.080 | 0.120 | 0.200 | 0.160 | 0.752 |
| 21 | 0.608 | 0.080 | 0.112 | 0.040 | 0.224 | 1.620 | 0.040 | 0.800 | 0.160 | 0.940 |
| 22 | 0.304 | 0.080 | 0.056 | 0.080 | 0.336 | 0.540 | 0.160 | 0.800 | 0.200 | 0.752 |
| 23 | 0.304 | 0.040 | 0.112 | 0.120 | 0.112 | 2.160 | 0.160 | 1.000 | 0.160 | 0.564 |
| 24 | 0.152 | 0.080 | 0.168 | 0.040 | 0.448 | 2.160 | 0.200 | 0.800 | 0.120 | 0.940 |
| 25 | 0.304 | 0.120 | 0.056 | 0.160 | 0.448 | 2.700 | 0.160 | 0.600 | 0.200 | 0.188 |
1.3 Case study 2
The second case study is of two-way diode array detector (DAD) HPLC data of a small embedded peak, that of 3-hydroxypyridine, buried within a major peak (2-hydroxypyridine). The concentration of the embedded peak varies between 1 and 5% of the 2-hydroxypyridine, and a series of 14 chromatograms (including replicates) are recorded whose concentrations are given in Table 3.| Sample | Conc./mM |
|---|---|
| 1 | 0.0158 |
| 2 | 0.0158 |
| 3 | 0.0315 |
| 4 | 0.0315 |
| 5 | 0.0315 |
| 6 | 0.0473 |
| 7 | 0.0473 |
| 8 | 0.0473 |
| 9 | 0.0473 |
| 10 | 0.0631 |
| 11 | 0.0631 |
| 12 | 0.0631 |
| 13 | 0.0789 |
| 14 | 0.0789 |
The chromatogram was sampled every 1 s, and a 40 s portion of each chromatogram was selected to contain the peak cluster, and aligned to the major peak maximum. Fifty-one wavelengths between 230 and 350 nm (sampled at 2.4 nm intervals) were recorded. Hence a dataset of dimensions 14 × 40 × 51 was obtained, the aim being to use multimode calibration to determine the concentration of the minor component. Further experimental details are reported elsewhere.4
The dataset is available in ESI Table S3†. It is arranged so that each column corresponds to a wavelength and there are 14 successive blocks, each of 40 rows (corresponding to successive points in time). Horizontal lines are used to divide each block for clarity. The chromatograms have been aligned.
2 Calibration methods
We will illustrate the methods of Sections 2.1–2.4 with dataset A of case study 1, and the methods of Section 2.5 with case study 2.2.1 Univariate calibration
Mathematically a series of experiments can be performed to give
| x ≈ c.s |
A simple method for solving this equation is as follows:
| c′ .x ≈ c′.c .s |
| (c′.c)−1.c′. x ≈ (c′.c)−1. (c′.c).s |
where the ′ is the transpose as described in Appendix A1.

Many conventional texts use summations rather than matrices for
determination of regression equations, but both approaches are equivalent.
In Fig. 1, the absorbance of the spectra of
case study 1A at 336 nm is plotted against the concentration of pyrene
(Table 1). The graph is approximately
linear, and provides a best fit slope calculated by

so that

![[x with combining circumflex]](https://www.rsc.org/images/entities/i_char_0078_0302.gif) = 0.291 ĉ. Note the hat (^) symbol which
indicates a prediction. The results are presented in Table 4.
= 0.291 ĉ. Note the hat (^) symbol which
indicates a prediction. The results are presented in Table 4. | ||
| Fig. 1 Absorption at 336 nm against concentration of pyrene. | ||
| Concentration | Absorbance | Predicted absorbance | Residual |
|---|---|---|---|
| 0.456 | 0.161 | 0.133 | 0.028 |
| 0.456 | 0.176 | 0.133 | 0.043 |
| 0.152 | 0.102 | 0.044 | 0.058 |
| 0.760 | 0.184 | 0.221 | −0.037 |
| 0.760 | 0.231 | 0.221 | 0.010 |
| 0.608 | 0.171 | 0.176 | −0.006 |
| 0.760 | 0.183 | 0.221 | −0.039 |
| 0.456 | 0.160 | 0.133 | 0.027 |
| 0.304 | 0.126 | 0.088 | 0.038 |
| 0.608 | 0.186 | 0.177 | 0.009 |
| 0.608 | 0.146 | 0.177 | −0.031 |
| 0.152 | 0.064 | 0.044 | 0.020 |
| 0.608 | 0.139 | 0.177 | −0.038 |
| 0.456 | 0.110 | 0.133 | −0.023 |
| 0.760 | 0.202 | 0.221 | −0.019 |
| 0.152 | 0.087 | 0.044 | 0.043 |
| 0.152 | 0.076 | 0.044 | 0.032 |
| 0.304 | 0.104 | 0.088 | 0.016 |
| 0.152 | 0.120 | 0.044 | 0.076 |
| 0.456 | 0.125 | 0.133 | −0.008 |
| 0.608 | 0.173 | 0.177 | −0.004 |
| 0.304 | 0.092 | 0.088 | 0.004 |
| 0.304 | 0.135 | 0.088 | 0.046 |
| 0.760 | 0.212 | 0.221 | −0.009 |
| 0.304 | 0.142 | 0.088 | 0.054 |
The quality of prediction can be determined by the residuals (or errors)
i.e. the difference between the observed and predicted,
i.e.x
−
; the less this is the
better. Generally the root mean error is calculated,

This error can be represented as a percentage of the mean E% = 100 (E/
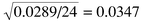
![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) ) = 24.1% in this case. It is always useful
to check the original graph (Fig. 1) just to
be sure, which appears a reasonable answer. Note that classical calibration
is slightly illogical in analytical chemistry. The aim of calibration is to
determine concentrations from spectral intensities, and not vice
versa yet the calibration equation in this section involves fitting a
model to determine a peak height from a known concentration.
) = 24.1% in this case. It is always useful
to check the original graph (Fig. 1) just to
be sure, which appears a reasonable answer. Note that classical calibration
is slightly illogical in analytical chemistry. The aim of calibration is to
determine concentrations from spectral intensities, and not vice
versa yet the calibration equation in this section involves fitting a
model to determine a peak height from a known concentration.For a new or unknown sample, the concentration can be estimated
(approximately) by using the inverse of the slope or
| ĉ = 3.44 x |
 | ||
| Fig. 2 Spectrum of pyrene superimposed over the spectra of the other pure PAHs. | ||
 | ||
| Fig. 3 Errors in (a) Classical and (b) Inverse calibration. | ||
Calibration can be performed by the inverse method where
| c ≈ x.b |
giving for this example, ĉ= 3.262 x. Note that b is only approximately the inverse of s (see above), because each model makes different assumptions about error distributions. However, for good data, both models should provide fairly similar predictions, if not there could be some other factor that influences the data, such as an intercept, non-linearities, outliers or unexpected noise distributions. For heteroscedastic noise distributions24 there are a variety of enhancements to linear calibration. However, these are rarely taken into consideration when extending the principles to the multivariate calibration.

Most chemometricians prefer inverse methods, but most traditional analytical chemistry texts introduce the classical approach to calibration. It is important to recognise that there are substantial differences in terminology in the literature, the most common problem being the distinction between ‘x’ and ‘y’ variables. In many areas of analytical chemistry, concentration is denoted by ‘x’, the response (such as a spectroscopic peak height) by ‘y’. However, most workers in the area of multivariate calibration have first been introduced to regression methods via spectroscopy or chromatography whereby the experimental data matrix is denoted as ‘X’, and the concentrations or predicted variables by ‘y’. In this paper we indicate the experimentally observed responses by ‘x’ such as spectroscopic absorbances of chromatographic peak areas, but do not use ‘y’ in order to avoid confusion.
| c ≈ b0+ b1x |
| c ≈ X . b |
| 1 | 0.456 |
| 1 | 0.456 |
| 1 | 0.152 |
| 1 | 0.760 |
| 1 | 0.760 |
| 1 | 0.608 |
| 1 | 0.760 |
| 1 | 0.456 |
| 1 | 0.304 |
| 1 | 0.608 |
| 1 | 0.608 |
| 1 | 0.152 |
| 1 | 0.608 |
| 1 | 0.456 |
| 1 | 0.760 |
| 1 | 0.152 |
| 1 | 0.152 |
| 1 | 0.304 |
| 1 | 0.152 |
| 1 | 0.456 |
| 1 | 0.608 |
| 1 | 0.304 |
| 1 | 0.304 |
| 1 | 0.760 |
| 1 | 0.304 |
Exactly the same principles can be employed for calculating the
coefficients as in Section 2.1.2, but in this case b is a vector
rather than scalar, and X is a matrix rather than a vector so
that
| b = (X′.X)−1.X′ . c |
| ĉ = −0.178 + 4.391 x |
The predicted concentrations are easy to obtain, the easiest approach
involving the use of matrix-based methods, so that
| ĉ = X.b |
representing an E% of 23.3%. Notice that, strictly speaking, the error term is divided by 23 (number of degrees of freedom rather than 25) to reflect the two parameters used in the model.

An alternative, and common, method for including the intercept is to
mean centre both the x and the c variables to fit the
equation
c
−
![[c with combining macron]](https://www.rsc.org/images/entities/b_i_char_0063_0304.gif) = (x
− = (x
−
![[x with combining macron]](https://www.rsc.org/images/entities/b_i_char_0078_0304.gif) )b )b |
| cenc = cenx b |
It is easy to show algebraically that the value of b is identical with b1 obtained for the uncentred data (=4.391 in this example), but includes the intercept, whereas the old value of b0 is given by (
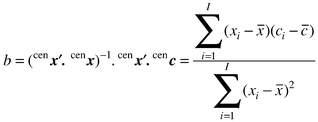
![[c with combining macron]](https://www.rsc.org/images/entities/i_char_0063_0304.gif) −b1), so the two methods are
related. It is common to centre both sets of variables for this reason, the
calculations being mathematically simpler than including an intercept term.
Note that the concentrations must be centred at the same time as the
response, and the predictions are of the concentrations minus their
mean.
−b1), so the two methods are
related. It is common to centre both sets of variables for this reason, the
calculations being mathematically simpler than including an intercept term.
Note that the concentrations must be centred at the same time as the
response, and the predictions are of the concentrations minus their
mean.It should be pointed out that the predictions for both methods described in this section differ from those obtained for the uncentred data. It is also useful to realise that it is also possible to use an intercept in models obtained using classical calibration; the details have been omitted in this section for brevity.
2.2 Multiple linear regression
In case study 1, only certain compounds absorb above 330 nm, the main ones being pyrene, fluoranthene, acenaphthylene and benzanthracene (note that the small absorbance due to a fifth component may be regarded as an interferent, although including this in the model will, of course, result in better predictions). It is possible to choose four wavelengths, preferably ones in which the absorbance ratios of these four compounds differ. In Fig. 4, the wavelengths 331, 335, 341 and 349 nm are indicated, and chosen for calibration.
![Spectra of pyrene, fluoranthene, acenaphthalene and
benzo[a])anthracene between 330 and 350 nm with 331, 335, 341 and
349 nm indicated.](/image/article/2000/AN/b003805i/b003805i-f4.gif) | ||
| Fig. 4 Spectra of pyrene, fluoranthene, acenaphthalene and benzo[a])anthracene between 330 and 350 nm with 331, 335, 341 and 349 nm indicated. | ||
Calibration equations can be obtained, as follows, using inverse
methods. First, select the absorbances of the 25 spectra at these four
wavelengths to give an X matrix with four columns and 25 rows.
Second, obtain the corresponding C matrix consisting of the
relevant concentrations (Table 6). The
aim is to find coefficients B relating X and C
by
| C ≈ X . B |
| B = (X′.X)−1.X′ .C |
| estimated [pyrene] = −1.827 A331 + 7.512 A335 −6.094 A341 + 2.355 A349 |
| C | X | ||||||
|---|---|---|---|---|---|---|---|
| 331 | 335 | 341 | 349 | Py | Ace | Benz | Fluora |
| 0.138 | 0.165 | 0.102 | 0.058 | 0.456 | 0.120 | 1.620 | 0.120 |
| 0.154 | 0.178 | 0.133 | 0.078 | 0.456 | 0.040 | 2.700 | 0.120 |
| 0.093 | 0.102 | 0.087 | 0.053 | 0.152 | 0.200 | 1.620 | 0.080 |
| 0.152 | 0.191 | 0.093 | 0.046 | 0.760 | 0.200 | 1.080 | 0.160 |
| 0.191 | 0.239 | 0.131 | 0.073 | 0.760 | 0.160 | 2.160 | 0.160 |
| 0.148 | 0.178 | 0.105 | 0.056 | 0.608 | 0.200 | 2.160 | 0.040 |
| 0.149 | 0.193 | 0.074 | 0.029 | 0.760 | 0.120 | 0.540 | 0.160 |
| 0.137 | 0.164 | 0.105 | 0.057 | 0.456 | 0.080 | 2.160 | 0.120 |
| 0.107 | 0.129 | 0.093 | 0.057 | 0.304 | 0.160 | 1.620 | 0.200 |
| 0.168 | 0.193 | 0.124 | 0.067 | 0.608 | 0.160 | 2.700 | 0.040 |
| 0.119 | 0.154 | 0.058 | 0.021 | 0.608 | 0.040 | 0.540 | 0.040 |
| 0.06 | 0.065 | 0.049 | 0.028 | 0.152 | 0.160 | 0.540 | 0.080 |
| 0.112 | 0.144 | 0.067 | 0.033 | 0.608 | 0.120 | 1.080 | 0.040 |
| 0.093 | 0.114 | 0.056 | 0.034 | 0.456 | 0.200 | 0.540 | 0.120 |
| 0.169 | 0.211 | 0.1 | 0.052 | 0.760 | 0.040 | 1.620 | 0.160 |
| 0.082 | 0.087 | 0.081 | 0.054 | 0.152 | 0.040 | 2.160 | 0.080 |
| 0.071 | 0.077 | 0.059 | 0.037 | 0.152 | 0.080 | 1.080 | 0.080 |
| 0.084 | 0.106 | 0.066 | 0.037 | 0.304 | 0.040 | 1.080 | 0.200 |
| 0.113 | 0.119 | 0.115 | 0.078 | 0.152 | 0.120 | 2.700 | 0.080 |
| 0.106 | 0.13 | 0.073 | 0.042 | 0.456 | 0.160 | 1.080 | 0.120 |
| 0.151 | 0.182 | 0.091 | 0.043 | 0.608 | 0.080 | 1.620 | 0.040 |
| 0.08 | 0.095 | 0.056 | 0.035 | 0.304 | 0.080 | 0.540 | 0.200 |
| 0.128 | 0.138 | 0.114 | 0.071 | 0.304 | 0.200 | 2.700 | 0.200 |
| 0.177 | 0.219 | 0.132 | 0.078 | 0.760 | 0.080 | 2.700 | 0.160 |
| 0.133 | 0.147 | 0.109 | 0.066 | 0.304 | 0.120 | 2.160 | 0.200 |
| B | ||||||
|---|---|---|---|---|---|---|
| Py | Ace | Benz | Fluora | |||
| 331 | −1.827 | 5.950 | −0.591 | −1.741 | ||
| 335 | 7.512 | −3.105 | −11.209 | 2.785 | ||
| 341 | −6.094 | −4.061 | 48.280 | −6.409 | ||
| 349 | 2.355 | 3.972 | −19.343 | 9.734 | ||
| Ĉ | ||||||
|---|---|---|---|---|---|---|
| Py | Ace | Benz | Fluora | |||
| 0.502 | 0.125 | 1.872 | 0.130 | |||
| 0.429 | 0.133 | 2.826 | 0.134 | |||
| 0.191 | 0.094 | 1.977 | 0.080 | |||
| 0.699 | 0.116 | 1.370 | 0.119 | |||
| 0.820 | 0.152 | 2.121 | 0.204 | |||
| 0.559 | 0.124 | 1.904 | 0.110 | |||
| 0.795 | 0.102 | 0.760 | 0.086 | |||
| 0.476 | 0.106 | 2.048 | 0.100 | |||
| 0.341 | 0.085 | 1.878 | 0.132 | |||
| 0.545 | 0.163 | 2.428 | 0.102 | |||
| 0.635 | 0.078 | 0.598 | 0.054 | |||
| 0.146 | 0.067 | 1.060 | 0.035 | |||
| 0.547 | 0.078 | 0.916 | 0.098 | |||
| 0.425 | 0.107 | 0.713 | 0.128 | |||
| 0.789 | 0.151 | 1.357 | 0.159 | |||
| 0.137 | 0.103 | 1.843 | 0.106 | |||
| 0.176 | 0.091 | 1.228 | 0.073 | |||
| 0.328 | 0.050 | 1.233 | 0.086 | |||
| 0.170 | 0.146 | 2.643 | 0.157 | |||
| 0.437 | 0.097 | 1.192 | 0.118 | |||
| 0.638 | 0.135 | 1.433 | 0.079 | |||
| 0.309 | 0.093 | 0.915 | 0.107 | |||
| 0.275 | 0.152 | 2.508 | 0.122 | |||
| 0.701 | 0.147 | 2.305 | 0.215 | |||
| 0.352 | 0.154 | 2.260 | 0.122 | |||
An estimated concentration matrix can be obtained by
| Ĉ = X . B |
(note that the divisor is 21 not 25 as four degrees of freedom are lost because there are four compounds in the model), equal to 0.042 or 9.34%, of the average concentration of pyrene, a significant improvement over the univariate model. Even further improvement could be obtained by including the intercept (usually performed by centring the data) and including the concentrations of more compounds.

It is possible also to employ classical methods. For the single
detector, single wavelength model of Section 2.1.1
| ĉ = x/s |
Ĉ =
![[X with combining circumflex]](https://www.rsc.org/images/entities/b_i_char_0058_0302.gif) .S′.(S.S′)−1 .S′.(S.S′)−1
|
| B ≈ Ŝ′. (Ŝ.Ŝ′)−1 |
Such equations make assumptions that the main analytes are all known, and work well only if this is true. Applying to mixtures where there are unknown interferents can result in serious estimation errors.
There is a fairly confusing literature on the use of multiple linear regression for calibration in chemometrics, primarily because many workers present their arguments in a very formalised manner. However, the choice and applicability of method depends on three main factors:(1) the number of compounds in the mixture (ten in this case) or responses to be estimated; (2) the number of experiments (25 in this case) often spectra or chromatograms; and (3) the number of detectors (131 wavelengths in this case).
In order to have a sensible model, the number of compounds must be less than or equal to the smaller of the number of experiments or number of detectors. In certain specialised cases this limitation can be infringed if it is known that there are correlations between the concentrations of different compounds. This may happen, for example, in environmental chemistry where there could be tens or hundreds of compounds in a sample, but the presence of one (e.g. a homologous series) suggests the presence of another, so, in practice there are only a few independent factors or groups of compounds. Also, correlations can be built into the design of a training set as discussed in Section 3.4. In most real-world situations there definitely will be correlations in complex multicomponent mixtures. However, the methods described below are for the case where the number of compounds is smaller than the number of experiments or number of detectors, for reasons described above.
The X data matrix is ideally related to the concentration and
spectral matrices by
| X = C . S |
| Ŝ = (C′.C)−1.C.X |
| Ĉ = (X.Ŝ.′(Ŝ.Ŝ′)−1 |
| Polyarene conc./mg L−1 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Spectrum | Py | Ace | Anth | Acy | Chry | Benz | Fluora | Fluore | Nap | Phen |
| 1 | 0.485 | 0.110 | 0.178 | 0.154 | 0.374 | 1.653 | 0.157 | 0.536 | 0.107 | 0.511 |
| 2 | 0.411 | 0.028 | 0.325 | 0.133 | 0.495 | 2.726 | 0.152 | 0.373 | 0.137 | 0.620 |
| 3 | 0.178 | 0.158 | 0.274 | 0.220 | 0.546 | 1.668 | 0.057 | 0.860 | 0.164 | 0.205 |
| 4 | 0.699 | 0.177 | 0.241 | 0.150 | 0.362 | 1.107 | 0.124 | 0.730 | 0.031 | 0.690 |
| 5 | 0.819 | 0.140 | 0.287 | 0.158 | 0.223 | 2.121 | 0.172 | 0.239 | 0.191 | 0.516 |
| 6 | 0.596 | 0.224 | 0.157 | 0.052 | 0.426 | 2.202 | 0.057 | 0.927 | 0.132 | 1.025 |
| 7 | 0.782 | 0.146 | 0.126 | 0.128 | 0.484 | 0.467 | 0.186 | 0.474 | 0.157 | 0.141 |
| 8 | 0.447 | 0.098 | 0.202 | 0.249 | 0.032 | 2.192 | 0.160 | 1.260 | 0.099 | 0.304 |
| 9 | 0.328 | 0.165 | 0.237 | 0.018 | 0.453 | 1.593 | 0.208 | 0.087 | 0.001 | 0.341 |
| 10 | 0.586 | 0.232 | 0.044 | 0.094 | 0.355 | 2.681 | 0.089 | 0.114 | 0.072 | 0.223 |
| 11 | 0.623 | 0.057 | 0.207 | 0.111 | 0.581 | 0.475 | 0.052 | 0.369 | 0.027 | 0.611 |
| 12 | 0.141 | 0.167 | 0.185 | 0.157 | 0.103 | 0.531 | 0.112 | 0.279 | 0.119 | 0.715 |
| 13 | 0.596 | 0.095 | 0.239 | 0.123 | 0.063 | 1.127 | -0.058 | 0.631 | 0.176 | 0.494 |
| 14 | 0.453 | 0.211 | 0.081 | 0.013 | 0.259 | 0.542 | 0.165 | 0.753 | 0.105 | 0.262 |
| 15 | 0.781 | 0.036 | 0.048 | 0.112 | 0.103 | 1.659 | 0.181 | 0.425 | 0.077 | 0.964 |
| 16 | 0.129 | 0.065 | 0.112 | 0.016 | 0.347 | 2.166 | 0.113 | 0.378 | 0.228 | 0.353 |
| 17 | 0.168 | 0.114 | 0.070 | 0.066 | 0.474 | 1.031 | 0.137 | 0.876 | 0.065 | 0.496 |
| 18 | 0.287 | 0.079 | 0.148 | 0.108 | 0.217 | 1.101 | 0.189 | 0.332 | 0.136 | 0.245 |
| 19 | 0.181 | 0.141 | 0.229 | 0.054 | 0.264 | 2.615 | 0.071 | 0.373 | 0.011 | 0.876 |
| 20 | 0.424 | 0.154 | 0.095 | 0.147 | 0.494 | 1.115 | 0.105 | 0.349 | 0.241 | 1.022 |
| 21 | 0.648 | 0.045 | 0.121 | 0.220 | 0.221 | 1.596 | -0.008 | 0.903 | 0.181 | 0.710 |
| 22 | 0.293 | 0.124 | 0.271 | 0.048 | 0.344 | 0.533 | 0.235 | 1.019 | 0.160 | 0.986 |
| 23 | 0.289 | 0.191 | 0.110 | 0.085 | 0.143 | 2.653 | 0.187 | 0.769 | 0.154 | 0.592 |
| 24 | 0.738 | 0.042 | 0.192 | 0.006 | 0.554 | 2.704 | 0.129 | 1.063 | 0.111 | 0.316 |
| 25 | 0.327 | 0.057 | 0.010 | 0.355 | 0.487 | 2.216 | 0.081 | 0.791 | 0.131 | 0.893 |
| E% | 7.88 | 32.86 | 15.61 | 59.93 | 13.43 | 3.23 | 46.24 | 23.21 | 29.41 | 16.52 |
 | ||
| Fig. 5 Spectra as predicted by MLR. | ||
MLR predicts concentrations well in this case because all significant compounds are included in the model, and so the data are almost completely modelled. If we knew of only a few compounds, there would be much poorer predictions. Consider the situation in which only pyrene, acenaphthene and anthracene are known. The C matrix now has only three columns, and the predicted concentrations are given in Table 8. The errors are, as expected, much larger than those of Table 7. The absorbances of the remaining seven compounds are mixed up with those of the three modelled components. This problem could be overcome if some characteristic wavelengths or regions of the spectrum at which the selected compounds absorb most strongly (see Section 2.2.2) are identified, or if the experiments were designed so that there are correlations in the data, or even by a number of methods for weighted regression, but the need to model all significant absorbants is a major limitation of MLR.
| Polyarene conc./mg L−1 | |||
|---|---|---|---|
| Spectrum | Py | Ace | Anth |
| 1 | 0.542 | 0.145 | 0.155 |
| 2 | 0.401 | 0.182 | 0.333 |
| 3 | 0.226 | 0.269 | 0.128 |
| 4 | 0.759 | 0.015 | 0.229 |
| 5 | 0.750 | 0.104 | 0.209 |
| 6 | 0.483 | 0.168 | 0.283 |
| 7 | 0.874 | 0.053 | 0.000 |
| 8 | 0.468 | 0.251 | 0.084 |
| 9 | 0.335 | 0.130 | 0.212 |
| 10 | 0.479 | 0.366 | −0.054 |
| 11 | 0.743 | −0.082 | 0.232 |
| 12 | 0.213 | 0.013 | 0.227 |
| 13 | 0.458 | −0.004 | 0.208 |
| 14 | 0.432 | 0.090 | 0.053 |
| 15 | 0.823 | 0.013 | 0.188 |
| 16 | 0.021 | 0.262 | 0.148 |
| 17 | 0.258 | 0.160 | 0.125 |
| 18 | 0.333 | 0.116 | 0.101 |
| 19 | 0.091 | 0.190 | 0.345 |
| 20 | 0.503 | 0.082 | 0.221 |
| 21 | 0.653 | 0.098 | 0.137 |
| 22 | 0.368 | −0.071 | 0.425 |
| 23 | 0.190 | 0.324 | 0.140 |
| 24 | 0.616 | 0.228 | 0.175 |
| 25 | 0.562 | 0.306 | 0.054 |
| E% | 28.01 | 115.74 | 61.89 |
The approach described above is related to classical calibration, but it
is also possible to envisage an inverse calibration model since
| Ĉ = X . B |
| B = (X′.X)−1.X′ .C |
Condition 2 either requires a large number of extra experiments or a reduction to 25 wavelengths. There have been a number of algorithms that have been developed to reduce the wavelengths to the most significant ones, so enabling inverse models to be used, but there is no real advantage over classical models unless very specific information is available about error distributions.
2.3 Principal components regression
MLR-based methods have the disadvantage that all significant components must be known. PCA (principal components analysis)-based methods do not require details of all components, although it is necessary to make a sensible estimate of how many significant components characterise a mixture, but not necessarily their chemical identities.PCA decomposes an X matrix into two smaller matrices, one of
scores (T) and the other of loadings (P) as
follows
| X = T . P |
 | ||
| Fig. 6 Principles of PCA. | ||
The scores matrix has the following properties: 1. The number of rows equals the number of rows in the original data matrix, usually the number of samples. 2. The number of columns equals the number of significant factors in the data, and can be any number from 1 upwards. Ideally it equals the number of compounds in the original dataset but noise and spectral similarity combine to distort this number. Each column corresponds to a principal component. 3. The sum of squares of the elements of each column of the scores matrix relates to a number called the eigenvalue, and is often given as a definition of the eigenvalue. The larger the eigenvalue the more significant the component. The principal components are calculated in order of significance.
The loadings matrix has the following properties: 1. The number of columns equals the number of columns in the original data matrix, usually the number of detectors, or wavelengths in this case study. 2. The number of rows equals the number of significant factors in the data. Each row corresponds to a principal component. 3. The sum of squares of the elements of each column equals 1.
Hence each principal component, a, is characterised by: (1) a
scores vector ta being the
ath column of T, (2) a loadings vector
pa being the ath
row of P; and (3) an eigenvalue
ga which may be defined by

The sum of eigenvalues over all significant components should equal approximately the sum of squares of the original data, and will never be more than this number.
Principal components (PCs) are often presented geometrically. Spectra can be represented as points in J -dimensional space where each of the J -axes represents the intensity at each wavelength. Hence in case study 1, each spectrum an be represented by a point in 131-dimensional space The dataset can be represented by 25 such points, and the pattern formed in this new space indicates information about the data.
The first PC can be defined as the best fit straight line in this multi-dimensional space. The scores represent the distance along this line, and the loadings the direction (angle) of the straight line. If there is only one compound in a series of spectra, all the spectra will fall approximately on the straight line, since the intensity of each spectrum will relate directly to concentration. This distance is the score of the PC. If there are two components, ideally two PCs will be calculated, and representing the axes of a plane. For ten compounds, ideally ten PCs are calculated to give a ten-dimensional subspace of the original 131 dimensional space (in this case).
Another important property of PCs is often loosely called
orthogonality. Numerically this means that

or ta. tb = 0 and pa. pb = 0 for two components a and b using vector notation. Some authors state that principal components are uncorrelated. Strictly speaking this property depends on data preprocessing, and is only true if the variables have been centred (down each column) prior to PCA. We will, however, use the terminology ‘orthogonality’ to refer to these properties below.

PCA can be used to reduce the number of original variables to a few reduced variables or PCs, by keeping only the largest or most significant PCs; methods for selecting how many components to keep are discussed in Section 3. In case study 1 an ideal situation would be to reduce the 131 wavelengths to ten PCs. There are a variety of methods of data preprocessing or scaling (such as centring and standardisation) that are sometimes used,20 but below we use the raw data directly. The scores of the first ten PCs are given in Table 9 . Using ten PCs implies that up to ten distinct compounds are in the mixture, but, unlike in MLR it is not necessary to know the concentrations of all these components in advance, only those of the calibrants. This property, of course, allows chemometric techniques to be employed in situations where only one or two compounds are of interest, for example measuring the concentration of chlorophyll in pigment extracts of plants, or the concentration of a nutrient in a food sample. There may be a dozen or more chemical components in the mixture, most of which are unknown or of no interest. Hence it is desired only to calibrate against the known compound.
| Spectrum | PC1 | PC2 | PC3 | PC4 | PC5 | PC6 | PC7 | PC8 | PC9 | PC10 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 6.066 | 0.032 | 0.098 | −0.002 | 0.048 | 0.037 | 0.011 | 0.003 | 0.033 | −0.003 |
| 2 | 8.040 | −0.155 | −0.481 | −0.001 | 0.069 | 0.007 | −0.051 | 0.016 | 0.077 | −0.005 |
| 3 | 6.261 | −0.064 | 0.261 | −0.212 | −0.373 | 0.086 | −0.080 | 0.043 | 0.025 | −0.015 |
| 4 | 5.877 | 0.606 | 0.119 | 0.061 | 0.117 | 0.120 | −0.007 | −0.012 | −0.031 | −0.026 |
| 5 | 6.928 | 0.072 | 0.012 | 0.399 | 0.164 | 0.009 | −0.009 | 0.069 | 0.037 | 0.016 |
| 6 | 7.587 | 0.101 | −0.188 | −0.075 | −0.042 | −0.044 | −0.017 | −0.026 | −0.096 | 0.009 |
| 7 | 4.320 | 0.373 | 0.667 | −0.148 | 0.214 | 0.002 | 0.023 | 0.073 | 0.008 | 0.010 |
| 8 | 6.491 | −0.290 | 0.302 | 0.296 | −0.161 | 0.026 | 0.035 | −0.023 | 0.024 | −0.080 |
| 9 | 5.651 | −0.117 | −0.295 | −0.145 | 0.182 | 0.166 | 0.018 | 0.014 | 0.020 | 0.013 |
| 10 | 6.657 | −0.979 | 0.360 | 0.053 | 0.157 | 0.090 | −0.005 | 0.022 | −0.060 | 0.041 |
| 11 | 4.442 | 0.845 | 0.051 | −0.209 | 0.226 | 0.055 | −0.072 | −0.037 | 0.005 | 0.015 |
| 12 | 3.612 | 0.542 | −0.083 | 0.213 | −0.265 | 0.092 | 0.045 | 0.020 | 0.000 | 0.021 |
| 13 | 4.144 | 0.493 | 0.005 | 0.354 | −0.119 | −0.077 | −0.100 | 0.042 | −0.039 | −0.003 |
| 14 | 3.657 | 0.163 | 0.287 | −0.152 | 0.014 | 0.000 | 0.071 | 0.057 | −0.051 | −0.021 |
| 15 | 5.666 | 0.200 | −0.042 | 0.294 | 0.356 | −0.089 | 0.079 | −0.078 | 0.013 | 0.009 |
| 16 | 5.566 | −0.582 | −0.277 | −0.158 | −0.129 | −0.146 | 0.009 | 0.059 | 0.028 | 0.036 |
| 17 | 4.775 | 0.039 | 0.067 | −0.412 | −0.087 | 0.001 | 0.042 | −0.026 | −0.009 | −0.022 |
| 18 | 4.174 | −0.034 | 0.069 | 0.035 | −0.011 | 0.000 | 0.049 | 0.040 | 0.047 | 0.007 |
| 19 | 7.023 | −0.269 | −0.691 | 0.090 | −0.057 | 0.104 | −0.027 | −0.046 | −0.036 | 0.008 |
| 20 | 5.735 | 0.458 | 0.073 | −0.105 | −0.130 | −0.083 | 0.021 | −0.001 | −0.002 | 0.078 |
| 21 | 5.620 | 0.277 | 0.297 | 0.190 | −0.071 | −0.118 | −0.059 | −0.019 | −0.012 | −0.011 |
| 22 | 5.266 | 0.999 | −0.461 | −0.158 | −0.137 | −0.048 | 0.081 | 0.013 | 0.004 | −0.026 |
| 23 | 7.060 | −0.677 | −0.117 | 0.115 | −0.143 | −0.025 | 0.076 | 0.011 | −0.037 | −0.009 |
| 24 | 7.805 | −0.411 | −0.118 | −0.289 | 0.293 | −0.129 | −0.056 | 0.007 | −0.005 | −0.055 |
| 25 | 7.332 | −0.243 | 0.523 | −0.076 | −0.193 | −0.009 | −0.005 | −0.138 | 0.045 | 0.028 |
If cn is a vector containing the
known concentration of compound n in the spectra (25 in this
instance), then the PC scores can be related as follows:
| cn ≈ T . rn |
| rn = (T′. T)−1. T′. cn |
| 0.209 |
| 0.309 |
| 0.291 |
| 0.830 |
| −0.517 |
| −0.395 |
| 0.878 |
| −1.229 |
| −0.363 |
There are a number of methods for determining how good the predictions are. Most use the calibration of predictions of concentration, on the c (or according to some authors y) block of data. These methods have been briefly introduced in the context of MLR, but when performing PCR there are a large number of methods for calculating errors, so we will expand on the techniques in this section.
The simplest method is to determine the sum of square of residuals
between the true and predicted concentrations

for compound n using a principal components. The larger this error, the worse the prediction, and the error reduces as more components are calculated.

Often the error is reported as a root mean square error

This error can also be reported as a percentage,
| E% = 100
E/n |
where n is the mean
concentration. It is also possible to report errors in terms of
quality of modelling of spectra (or chromatograms), often called the
x block error.
The quality of modelling of the spectra using PCA (the x
variance) can likewise be calculated as follows:

However, this error also can be expressed in terms of eigenvalues or scores, so that
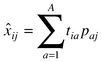
for A principal components.
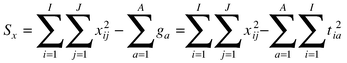
These can be converted to root mean square errors as above,
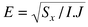
The percentage root mean square error may be defined by (for uncentred
data)
| E% = 100
E/ |
Note that if x is centred, the divisor is usually defined by
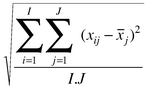 j is the
average of all the measurements for the samples for variable j:
obviously there are several other ways of defining this error; again each
investigator has his or her own favourites.
j is the
average of all the measurements for the samples for variable j:
obviously there are several other ways of defining this error; again each
investigator has his or her own favourites.Note that the x block error depends only on how many PCs have been used in the model, but the error in the c block depends also on the specific compound, there being a different percentage error for each compound in the mixture. For 0 PCs, the estimates of the PCs and concentrations are simply 0 (or the mean if the data have been centred). The graphs of errors for both the concentration estimates of pyrene and spectra as increasing numbers of PCs are calculated are given in Fig. 7. Although the x error graph declines steeply, which might falsely suggest only a small number of PCs are required for the model, the c error graph exhibits a much gentler decline. Some chemometricians prefer to plot the graph of ‘variances’; these are the mean square error if the data have been centred, and these graphs are presented either as percentage variance remaining (or explained by each PC) or, for the x block, by eigenvalues. Fig. 8 shows how the prediction for pyrene for dataset A of case study 1 improves with increasing PCs.
 | ||
| Fig. 7 Error for PCR estimates of pyrene as increasing number of components are employed. | ||
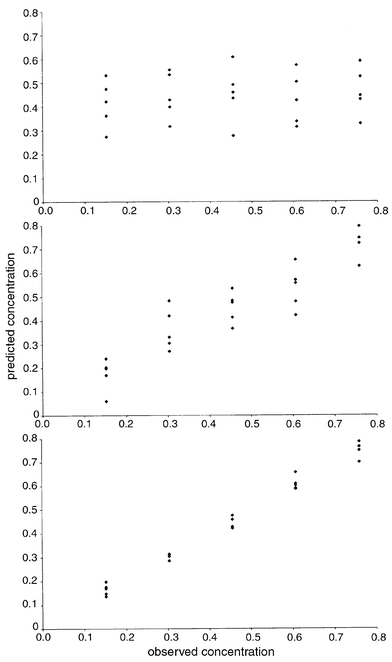 | ||
| Fig. 8 Predicted concentrations for pyrene using PCR as one, five and ten principal components are calculated. | ||
If the concentration of some or all the compounds are known PCR can be
extended simply by replacing the vector
ck with a matrix C, each
column corresponding to a compound in the mixture, so that
| C ≈ T . R |
| R = (T′. T)−1. T′. C |
| X= T.P = T.R.R.−1.P = Ĉ.Ŝ |
| Polyarene conc./mg L−1 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Spectrum | Py | Ace | Anth | Acy | Chry | Benz | Fluora | Fluore | Nap | Phen |
| 1 | 0.475 | 0.108 | 0.185 | 0.150 | 0.374 | 1.644 | 0.156 | 0.532 | 0.110 | 0.494 |
| 2 | 0.422 | 0.054 | 0.322 | 0.132 | 0.465 | 2.712 | 0.161 | 0.473 | 0.146 | 0.631 |
| 3 | 0.170 | 0.175 | 0.274 | 0.191 | 0.559 | 1.657 | 0.081 | 0.824 | 0.153 | 0.207 |
| 4 | 0.700 | 0.178 | 0.244 | 0.160 | 0.346 | 1.114 | 0.126 | 0.788 | 0.050 | 0.678 |
| 5 | 0.803 | 0.131 | 0.283 | 0.163 | 0.214 | 2.132 | 0.163 | 0.279 | 0.188 | 0.537 |
| 6 | 0.601 | 0.201 | 0.162 | 0.085 | 0.424 | 2.214 | 0.073 | 0.951 | 0.139 | 1.003 |
| 7 | 0.786 | 0.144 | 0.115 | 0.140 | 0.466 | 0.482 | 0.150 | 0.520 | 0.158 | 0.185 |
| 8 | 0.428 | 0.118 | 0.193 | 0.195 | 0.066 | 2.160 | 0.167 | 1.103 | 0.078 | 0.319 |
| 9 | 0.311 | 0.122 | 0.202 | 0.107 | 0.410 | 1.654 | 0.148 | 0.190 | 0.034 | 0.467 |
| 10 | 0.590 | 0.213 | 0.047 | 0.120 | 0.332 | 2.701 | 0.079 | 0.219 | 0.087 | 0.222 |
| 11 | 0.610 | 0.077 | 0.191 | 0.109 | 0.571 | 0.488 | 0.072 | 0.393 | 0.046 | 0.671 |
| 12 | 0.147 | 0.158 | 0.203 | 0.139 | 0.110 | 0.523 | 0.107 | 0.288 | 0.108 | 0.654 |
| 13 | 0.587 | 0.116 | 0.240 | 0.086 | 0.099 | 1.120 | −0.011 | 0.537 | 0.160 | 0.490 |
| 14 | 0.459 | 0.165 | 0.077 | 0.075 | 0.239 | 0.565 | 0.119 | 0.793 | 0.114 | 0.269 |
| 15 | 0.765 | 0.030 | 0.055 | 0.094 | 0.118 | 1.653 | 0.179 | 0.373 | 0.072 | 0.934 |
| 16 | 0.136 | 0.058 | 0.102 | 0.037 | 0.351 | 2.146 | 0.103 | 0.320 | 0.223 | 0.389 |
| 17 | 0.176 | 0.102 | 0.075 | 0.087 | 0.465 | 1.021 | 0.126 | 0.883 | 0.072 | 0.468 |
| 18 | 0.285 | 0.075 | 0.132 | 0.111 | 0.218 | 1.106 | 0.151 | 0.294 | 0.129 | 0.301 |
| 19 | 0.198 | 0.141 | 0.229 | 0.084 | 0.253 | 2.626 | 0.072 | 0.415 | 0.034 | 0.878 |
| 20 | 0.421 | 0.142 | 0.114 | 0.122 | 0.513 | 1.108 | 0.120 | 0.327 | 0.221 | 0.960 |
| 21 | 0.657 | 0.094 | 0.159 | 0.130 | 0.267 | 1.541 | 0.061 | 0.804 | 0.158 | 0.582 |
| 22 | 0.313 | 0.109 | 0.253 | 0.088 | 0.331 | 0.541 | 0.187 | 1.008 | 0.158 | 1.032 |
| 23 | 0.312 | 0.171 | 0.109 | 0.103 | 0.141 | 2.661 | 0.148 | 0.771 | 0.142 | 0.582 |
| 24 | 0.750 | 0.049 | 0.170 | 0.052 | 0.528 | 2.723 | 0.118 | 1.115 | 0.126 | 0.385 |
| 25 | 0.304 | 0.094 | 0.045 | 0.224 | 0.556 | 2.181 | 0.140 | 0.626 | 0.090 | 0.776 |
| E% | 7.61 | 29.52 | 18.39 | 42.46 | 9.52 | 3.18 | 38.75 | 20.15 | 25.12 | 19.36 |
2.4 Partial least squares
PLS is often regarded as the major regression technique for multivariate data. In fact in many cases it is applied inappropriately and is not justified by the data. In areas outside mainstream analytical chemistry such as QSAR, or even biometrics and psychometrics, PLS certainly is an invaluable tool, because the underlying factors have little or no physical meaning so a linearly additive model in which each underlying factor can be interpreted chemically is not expected. In spectroscopy or chromatography we usually expect linear additivity, and this is especially the case in analytical chemistry calibration. Nevertheless, PLS can be a useful tool when there is partial knowledge of the data, an excellent example being the measurement of protein in wheat by NIR spectroscopy.6,7 Under such conditions, the model will be obtained from a series of wheat samples, and PLS will try to use typical features in this dataset to establish a relationship with the known amount of protein. Unlike MLR it does not require an exact model of all components in the data. PLS models can be very robust provided that future samples contain similar features to the original data, but predictions are essentially statistical. An example might be the determination of vitamin C in orange juices using spectroscopy: a very reliable PLS model could be obtained using orange juices from a particular region of Spain, but what if some Brazilian orange juice is included? There is no guarantee that the model will perform well on the new data, as there may be different spectral features. The originators of PLS are well aware of the shortcomings as well as the successes of the method, but it is important for the analytical chemist to be very alert to potential pitfalls.One important practical aspect of PLS is that it takes into account errors both in the concentration estimates and spectra. A method such as PCR will assume that the concentration estimates are error free. Much traditional statistics rest on this assumption, that all errors are in the dependent variables (spectra). If in medicine it is decided to determine the concentration of a compound in the urine of patients as a function of age, it is assumed that age can be estimated exactly, the statistical variation being in the concentration of a compound and the nature of the urine sample. Yet in chemistry there are often significant errors in sample preparation, for example, accuracy of weighings and dilutions and so the independent variable (c) in itself also contains errors. With modern spectrometers, these are sometimes larger than spectroscopic errors. One way of overcoming this difficulty is to try to minimise the covariance between both types of variables, namely the x (spectroscopic) and c (concentration) variables.
| X = T.P + E |
| c = T.q + f |
 | ||
| Fig. 9 Principles of PLS1. | ||
There are a number of alternative ways of presenting the PLS regression equations in the literature, all, in practice, equivalent. In the models above, there are three arrays T, P and q and a conventional analogy to PCA sets P as a matrix, each of whose rows has a sum of squares equal to 1. From this the magnitude of T follows, which determines q. Some packages calculate a vector proportional to q, which is also normalised, in analogy to a loadings vector. In such a situation, the second equation becomes a product of three arrays, the first one proportional to T, the second one a diagonal matrix consisting of scaling factors, and the third one a normalised vector proportional to q. It is also possible to convert both equations to products of three arrays, but the models used in this paper have the simplicity of a single scores matrix, with the disadvantage of a vector q that is not normalised.
For a dataset consisting of 25 spectra observed at 131 wavelengths, for which eight PLS components are calculated, there will be: a T matrix of dimensions 25 × 8; a P matrix of dimensions 8 × 131; an E matrix of dimensions 25 × 131; a q vector of dimensions 8 × 1 and an f vector of dimensions 25 × 1.
Each successive PLS component approximates both the concentration and spectral data better. For each component, there will be a: spectral scores vector t; spectral loadings vector p and concentration loadings scalar q.
The approximation to the concentration as successive PLS components are calculated is simply the sum of t.q for each successive component. This approach is possible in PLS1 because each successive component is orthogonal.
In case study 1, there are ten compounds, so it is possible to perform PLS1 separately on each of the ten compounds. In each case it is possible compute several PLS components, if 15 were calculated for each compound, there will be 150 PLS components in total.
In most implementations of PLS it is conventional to centre both the x and c data, by subtracting the mean of each column before analysis. In fact, there is no general scientific need to do this. Many spectroscopists and chromatographers perform PCA uncentred; however, many early applications of PLS (e.g. outside chemistry) were of such a nature that centring the data was appropriate. Much of the history of PLS in analytical chemistry relates to applications in NIR spectroscopy, where there are specific spectroscopic problems, such as due to baselines, which, in turn would favour centring. However, as generally applied to analytical chemistry, uncentred PLS is perfectly acceptable. Below, though, we review the most widespread implementation for the sake of compatibility with the most common computational implementations of the method.
For a given compound, the remaining percentage error in the x
matrix for A PLS components can be expressed in a variety of ways
(see Section 2.3). Note that there are slight differences according to
authors that take into account the number of degrees of freedom left in the
model. The predicted measurements simply involve calculating
= T.P and adding on the column averages where
appropriate, and error indicators in the x block can be expressed
identically with those for PCA and can be calculated, see Section 2.3.2.
The only difference is that each compound generates a separate scores
matrix, unlike PCR where there is a single scores matrix for all compounds
in the mixture.
The concentration is predicted by

| cn =
Tn qn+
n |
n is a
vector of the average concentration. Hence the scores of each PLS component
are proportional to the contribution of the component to the concentration
estimate. The method of the concentration estimation for two PLS components
and pyrene is presented in Table
12.
| Component 1 scores | q = 0.0607; conc. est. (ti1q) | Component 2 scores | q = 0.318; conc. est. (ti2q) | Centred conc. est. (ti1q + ti2q) |
|---|---|---|---|---|
| 0.333 | 0.020 | 0.127 | 0.040 | 0.060 |
| 1.999 | 0.121 | −0.301 | −0.096 | 0.026 |
| 0.147 | 0.009 | −0.352 | −0.112 | −0.103 |
| 0.570 | 0.035 | 0.775 | 0.246 | 0.281 |
| 1.504 | 0.091 | 0.529 | 0.168 | 0.259 |
| 1.743 | 0.106 | 0.011 | 0.004 | 0.109 |
| −0.881 | −0.053 | 0.869 | 0.276 | 0.223 |
| 0.679 | 0.041 | −0.020 | −0.006 | 0.035 |
| −0.428 | −0.026 | −0.370 | −0.118 | −0.144 |
| 0.659 | 0.040 | −0.389 | −0.124 | −0.084 |
| −0.894 | −0.054 | 0.759 | 0.241 | 0.187 |
| −2.335 | −0.142 | −0.091 | −0.029 | −0.171 |
| −1.511 | −0.092 | 0.277 | 0.088 | −0.004 |
| −2.159 | −0.131 | 0.021 | 0.007 | −0.124 |
| 0.305 | 0.019 | 0.605 | 0.192 | 0.211 |
| −1.028 | −0.062 | −1.109 | −0.352 | −0.415 |
| −1.364 | −0.083 | −0.402 | −0.128 | −0.211 |
| −1.813 | −0.110 | −0.242 | −0.077 | −0.187 |
| 0.601 | 0.037 | −0.833 | −0.265 | −0.228 |
| 0.032 | 0.002 | 0.247 | 0.079 | 0.080 |
| 0.130 | 0.008 | 0.484 | 0.154 | 0.162 |
| −0.544 | −0.033 | 0.184 | 0.058 | 0.025 |
| 0.728 | 0.044 | −0.765 | −0.243 | −0.199 |
| 1.933 | 0.117 | −0.124 | −0.039 | 0.078 |
| 1.592 | 0.097 | 0.110 | 0.035 | 0.132 |
The mean square error in the concentration estimate can be computed just as in PCR, although the value of ĉin will differ. It is also possible to provide a number of equivalent equations for this error using t and q which are left to the reader. In the case of the concentration estimates, it is usual to adjust the sum of squares according to the number of PLS components, because this number is often similar in magnitude to the number of objects in the dataset; for example, there are 25 spectra in case study 1, but we might want to look at the error when ten PLS components are calculated. These error terms will also be discussed in Section 3.1. Note an interesting difference between the conventional equations for errors in the x and c data blocks: in the former the mean is subtracted from the overall sum of squares since the data are usually mean-centred prior to PLS, whereas for the latter the raw data are usually used as the mean concentration is generally added back on to the data so predictions are expressed in the original concentration units.
These calculations are illustrated for pyrene. Table 13 is of the first 15 eigenvalues for PLS1
using pyrene as the calibrant. The total sum of squares of the mean centred
spectra is 50.522, hence the first two eigenvalues account for
100x(38.578+6.269)/50.522 = 88.77% of the overall sum of squares, giving a
root mean square error after two PLS components have been calculated
of

| 6.269 |
| 2.563 |
| 1.697 |
| 0.624 |
| 0.536 |
| 0.081 |
| 0.048 |
| 0.0146 |
| 0.0261 |
| 0.0247 |
| 0.0159 |
| 0.0094 |
| 0.0026 |
| 0.0056 |
Table 14 is of the concentration predictions using two components. The sum of squares of the errors is 0.376. Dividing this by 22 and taking the square root leads to a root mean square error of 0.131 mg L−1. The average concentration of pyrene is 0.456 mg L−1. Hence the percentage root mean square error is 28.81%.
| Prediction | Error |
|---|---|
| 0.516 | 0.060 |
| 0.482 | 0.026 |
| 0.353 | 0.201 |
| 0.737 | −0.023 |
| 0.715 | −0.045 |
| 0.565 | −0.043 |
| 0.679 | −0.081 |
| 0.491 | 0.035 |
| 0.312 | 0.008 |
| 0.372 | −0.236 |
| 0.643 | 0.035 |
| 0.285 | 0.133 |
| 0.452 | −0.156 |
| 0.332 | −0.124 |
| 0.667 | −0.093 |
| 0.041 | −0.111 |
| 0.245 | 0.093 |
| 0.269 | −0.035 |
| 0.228 | 0.076 |
| 0.536 | 0.080 |
| 0.618 | 0.010 |
| 0.481 | 0.177 |
| 0.257 | −0.047 |
| 0.534 | −0.226 |
| 0.588 | 0.284 |
It is important to recognise that the percentage error of prediction in concentration may be different to the percentage error of prediction of the original spectra.
The root mean square percentage errors for modelling both spectra and concentrations of pyrene are presented in Fig. 10. Often these are plotted using a logarithmic scale for clarity. Fourteen components are required to obtain an error of prediction of concentration of less than 1%, whereas only 21 are needed to reach this for the spectral data. It is important to notice that there is not a sharp cut-off at ten components. If the number of compounds in the mixture spectra are unknown, it would not be at all obvious how complex the mixture is. Below we will discuss methods for determining the optimum number of components. The prediction error for pyrene using PLS1 and ten significant components, in this case, is considerably better than that using PCR, 3.40% as opposed to 7.61%. However, these raw errors are not always very useful indicators.
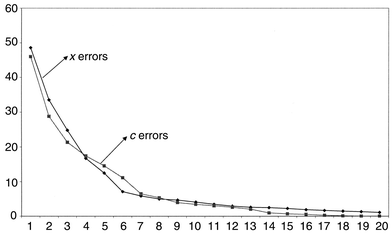 | ||
| Fig. 10 Root mean square errors for prediction of spectra and concentration of pyrene using PLS1 as successive number of components are employed. | ||
Fig. 11 represents the same data for acenaphthylene. Whereas the x block modelling error is fairly similar to that of pyrene, the concentration is modelled much less well, a consequence of the substantial spectral overlap and lack of significant features.
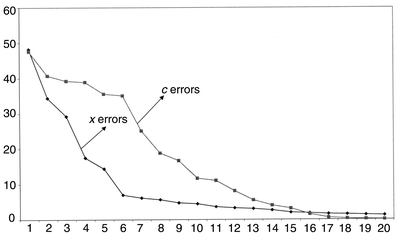 | ||
| Fig. 11 Root mean square errors for prediction of spectra and concentration of acenaphthylene using PLS1 as successive number of components are employed. | ||
The errors using ten PLS components are summarised in Table 15, and are better than PCR in this case. There is, however, an important philosophical consideration about what is a better prediction; although the measured c or concentration variables are obtained with greater accuracy, it is essential to recognise that there could be errors, in turn, in these concentration measurements, so PLS could simply be predicting poorer concentration estimates more accurately because the algorithm takes into account the c as well as x values. There is no easy answer.
| Polyarene conc./mg L−1 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Spectrum | Py | Ace | Anth | Acy | Chry | Benz | Fluora | Fluore | Nap | Phen |
| 1 | 0.438 | 0.133 | 0.158 | 0.123 | 0.340 | 1.636 | 0.118 | 0.614 | 0.116 | 0.601 |
| 2 | 0.462 | 0.043 | 0.282 | 0.210 | 0.447 | 2.709 | 0.116 | 0.382 | 0.153 | 0.749 |
| 3 | 0.155 | 0.195 | 0.280 | 0.162 | 0.559 | 1.623 | 0.083 | 0.813 | 0.160 | 0.187 |
| 4 | 0.729 | 0.183 | 0.219 | 0.195 | 0.336 | 1.108 | 0.115 | 0.781 | 0.042 | 0.761 |
| 5 | 0.788 | 0.170 | 0.279 | 0.114 | 0.222 | 2.119 | 0.165 | 0.182 | 0.169 | 0.548 |
| 6 | 0.608 | 0.211 | 0.175 | 0.059 | 0.452 | 2.168 | 0.055 | 0.811 | 0.116 | 0.931 |
| 7 | 0.760 | 0.113 | 0.119 | 0.168 | 0.439 | 0.552 | 0.176 | 0.620 | 0.197 | 0.174 |
| 8 | 0.471 | 0.086 | 0.229 | 0.174 | 0.114 | 2.124 | 0.129 | 0.985 | 0.038 | 0.180 |
| 9 | 0.305 | 0.158 | 0.230 | 0.033 | 0.449 | 1.611 | 0.194 | 0.180 | 0.022 | 0.370 |
| 10 | 0.605 | 0.169 | 0.050 | 0.159 | 0.334 | 2.732 | 0.053 | 0.200 | 0.084 | 0.210 |
| 11 | 0.625 | 0.028 | 0.228 | 0.130 | 0.575 | 0.512 | 0.051 | 0.402 | 0.037 | 0.548 |
| 12 | 0.155 | 0.156 | 0.179 | 0.189 | 0.099 | 0.539 | 0.095 | 0.289 | 0.119 | 0.736 |
| 13 | 0.591 | 0.115 | 0.275 | 0.045 | 0.122 | 1.094 | 0.030 | 0.560 | 0.151 | 0.388 |
| 14 | 0.471 | 0.203 | 0.060 | 0.051 | 0.232 | 0.526 | 0.125 | 0.779 | 0.084 | 0.351 |
| 15 | 0.755 | 0.038 | 0.057 | 0.081 | 0.113 | 1.630 | 0.155 | 0.415 | 0.073 | 0.938 |
| 16 | 0.148 | 0.026 | 0.114 | 0.038 | 0.340 | 2.167 | 0.058 | 0.399 | 0.193 | 0.364 |
| 17 | 0.157 | 0.094 | 0.050 | 0.115 | 0.447 | 1.047 | 0.072 | 0.973 | 0.081 | 0.573 |
| 18 | 0.296 | 0.058 | 0.157 | 0.139 | 0.218 | 1.100 | 0.191 | 0.381 | 0.140 | 0.220 |
| 19 | 0.151 | 0.118 | 0.221 | 0.088 | 0.219 | 2.695 | 0.088 | 0.613 | 0.056 | 0.936 |
| 20 | 0.460 | 0.159 | 0.115 | 0.101 | 0.552 | 1.075 | 0.123 | 0.194 | 0.192 | 0.935 |
| 21 | 0.609 | 0.080 | 0.111 | 0.188 | 0.216 | 1.615 | 0.041 | 1.015 | 0.203 | 0.762 |
| 22 | 0.305 | 0.092 | 0.272 | 0.079 | 0.336 | 0.563 | 0.211 | 0.980 | 0.169 | 0.962 |
| 23 | 0.303 | 0.179 | 0.117 | 0.134 | 0.122 | 2.693 | 0.205 | 0.794 | 0.188 | 0.550 |
| 24 | 0.756 | 0.076 | 0.170 | 0.036 | 0.551 | 2.691 | 0.166 | 1.049 | 0.130 | 0.378 |
| 25 | 0.297 | 0.118 | 0.053 | 0.189 | 0.566 | 2.171 | 0.183 | 0.589 | 0.083 | 0.750 |
| E% | 3.403 | 10.950 | 4.496 | 11.638 | 2.675 | 1.582 | 14.857 | 5.821 | 9.328 | 3.643 |
It is a simple extension to predict all the concentrations simultaneously, the PLS2 predictions, together with root mean square errors being given in Table 16. Note that there is now only one set of scores and loadings for the x (spectroscopic) dataset, and one set of eigenvalues common to all ten compounds. However, the concentration estimates are different when using PLS2 to PLS1. In this way PLS differs from PCR where it does not matter if each variable is modelled separately or all together. The reasons are rather complex but relate to the fact that for PCR the principal components are calculated independently of how many concentration variables are used in the regression; however, the PLS components are influenced by the concentration variable.
| Polyarene conc./mg L−1 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Spectrum | Py | Ace | Anth | Acy | Chry | Benz | Fluora | Fluore | Nap | Phen |
| 1 | 0.477 | 0.111 | 0.175 | 0.145 | 0.367 | 1.660 | 0.149 | 0.563 | 0.097 | 0.520 |
| 2 | 0.434 | 0.071 | 0.313 | 0.116 | 0.475 | 2.701 | 0.156 | 0.402 | 0.139 | 0.647 |
| 3 | 0.172 | 0.177 | 0.278 | 0.184 | 0.564 | 1.650 | 0.084 | 0.797 | 0.149 | 0.187 |
| 4 | 0.701 | 0.185 | 0.231 | 0.159 | 0.344 | 1.121 | 0.119 | 0.767 | 0.046 | 0.715 |
| 5 | 0.813 | 0.146 | 0.281 | 0.144 | 0.230 | 2.111 | 0.163 | 0.170 | 0.179 | 0.522 |
| 6 | 0.602 | 0.214 | 0.156 | 0.085 | 0.435 | 2.189 | 0.073 | 0.840 | 0.149 | 1.011 |
| 7 | 0.785 | 0.138 | 0.119 | 0.133 | 0.464 | 0.486 | 0.152 | 0.541 | 0.145 | 0.160 |
| 8 | 0.423 | 0.113 | 0.210 | 0.197 | 0.077 | 2.151 | 0.179 | 1.066 | 0.095 | 0.271 |
| 9 | 0.310 | 0.115 | 0.216 | 0.109 | 0.413 | 1.648 | 0.155 | 0.201 | 0.040 | 0.430 |
| 10 | 0.590 | 0.213 | 0.044 | 0.125 | 0.332 | 2.700 | 0.076 | 0.214 | 0.088 | 0.236 |
| 11 | 0.603 | 0.061 | 0.207 | 0.121 | 0.570 | 0.490 | 0.079 | 0.440 | 0.058 | 0.635 |
| 12 | 0.151 | 0.158 | 0.197 | 0.142 | 0.105 | 0.531 | 0.101 | 0.329 | 0.108 | 0.683 |
| 13 | 0.583 | 0.101 | 0.256 | 0.099 | 0.096 | 1.120 | −0.004 | 0.599 | 0.173 | 0.462 |
| 14 | 0.463 | 0.168 | 0.071 | 0.079 | 0.236 | 0.568 | 0.115 | 0.813 | 0.118 | 0.301 |
| 15 | 0.762 | 0.026 | 0.056 | 0.102 | 0.111 | 1.660 | 0.180 | 0.419 | 0.078 | 0.944 |
| 16 | 0.135 | 0.044 | 0.113 | 0.039 | 0.350 | 2.160 | 0.103 | 0.383 | 0.218 | 0.362 |
| 17 | 0.175 | 0.099 | 0.068 | 0.096 | 0.452 | 1.040 | 0.120 | 0.963 | 0.074 | 0.510 |
| 18 | 0.282 | 0.058 | 0.149 | 0.114 | 0.213 | 1.114 | 0.159 | 0.376 | 0.129 | 0.255 |
| 19 | 0.187 | 0.128 | 0.223 | 0.097 | 0.233 | 2.655 | 0.067 | 0.531 | 0.029 | 0.907 |
| 20 | 0.429 | 0.154 | 0.110 | 0.113 | 0.527 | 1.088 | 0.117 | 0.226 | 0.218 | 0.961 |
| 21 | 0.653 | 0.090 | 0.142 | 0.135 | 0.245 | 1.577 | 0.048 | 0.919 | 0.143 | 0.642 |
| 22 | 0.311 | 0.109 | 0.258 | 0.082 | 0.337 | 0.533 | 0.193 | 0.966 | 0.156 | 1.004 |
| 23 | 0.309 | 0.172 | 0.109 | 0.104 | 0.139 | 2.656 | 0.151 | 0.765 | 0.141 | 0.577 |
| 24 | 0.749 | 0.052 | 0.170 | 0.051 | 0.529 | 2.719 | 0.121 | 1.100 | 0.132 | 0.385 |
| 25 | 0.301 | 0.095 | 0.046 | 0.228 | 0.557 | 2.174 | 0.143 | 0.606 | 0.098 | 0.776 |
| E% | 7.398 | 26.813 | 12.068 | 42.827 | 7.495 | 2.711 | 36.921 | 10.768 | 30.415 | 13.640 |
In some cases PLS2 is helpful, especially since it is easier to perform computationally. Instead of obtaining ten independent models, one for each PAH, in this example, we can analyse all the data in one go. However, in many situations PLS2 concentration estimates are, in fact, worse than PLS1 estimates, so a good strategy might be to perform PLS2 as a first step, which could provide further information such as which wavelengths are significant and which concentrations can be determined with a high degree of confidence, and then perform PLS1 individually for the most appropriate compounds.
2.5 Multiway methods
Two way data such as DAD-HPLC, LC-MS and LC-NMR are increasingly common in analytical chemistry, especially with the growth of coupled chromatography. Conventionally either a univariate parameter (e.g. a peak area at a given wavelength) (methods of Section 2.1) or a chromatographic elution profile at a single wavelength (methods of Sections 2.2–2.4) is used for calibration, allowing the use of standard regression techniques described above. However, additional information has been recorded for each sample, often involving both an elution profile and a spectrum. A series of two way chromatograms are available, and can be organised into a three-way array often visualised as a box. Each level of the box consists of a single chromatogram. Sometimes these three-way arrays are called “tensors” but tensors often have special properties in physics which are unnecessarily complex and confusing to the chemometrician. We will refer to tensors only where it helps understand the existing methods.Enhancements of the standard methods for multivariate calibration are required. Although it is possible to use methods such as three- way MLR, most chemometricians have concentrated on developing approaches based on PLS, which we will be restricted to below. The data will be illustrated using case study 2.
 | ||
| Fig. 12 Unfolding a data matrix. | ||
It is now a simple task to perform PLS (or indeed any other multivariate approach), as discussed above. The 2040 variables are centred and the prediction of the concentration of 3-hydroxypyridine when three PLS components are employed is given in Fig. 13. The error of prediction of the concentration of 3-hydroxypyridine is presented in Fig. 14 for increasing number of components. Notice that several graphs could be produced of the effectiveness of the model, ranging from the eigenvalues (related to the x variables), to the percentage prediction error in the concentration variables, and the percentage of the chromatographic data modelled by each successive component. It is interesting that three PLS components appear to be required to give a good model, even though there are only two compounds in this region of the chromatogram (the major one and the impurity). There could be other factors such as noise that are modelled by these PLS components.
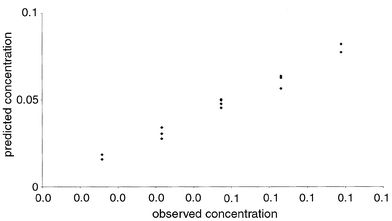 | ||
| Fig. 13 Predicted versus true concentrations of 3-hydroxypyridine (case study 2), using 3 PLS components and an unfolded data matrix as discussed in Section 2.5.1. | ||
 | ||
| Fig. 14 Error in response of the first 10 PLS components for the data discussed in Section 2.5.1. | ||
It is possible to improve the method by scaling the data, but it is important to be very careful to think about the consequences of the various methods employed. It is sometimes possible to scale first the two way data and then unfold. However, a final centring should normally be performed on the unfolded matrix. In addition, variable selection can have a significant influence on the effectiveness of unfolded PLS models, since not all the 2040 variables are going to be particularly relevant or informative.
Centring can be complex for three-way data, and there is no inherent reason to do this, therefore, for simplicity, in this section no centring is used, so raw concentrations and chromatographic/spectroscopic measurements are employed.
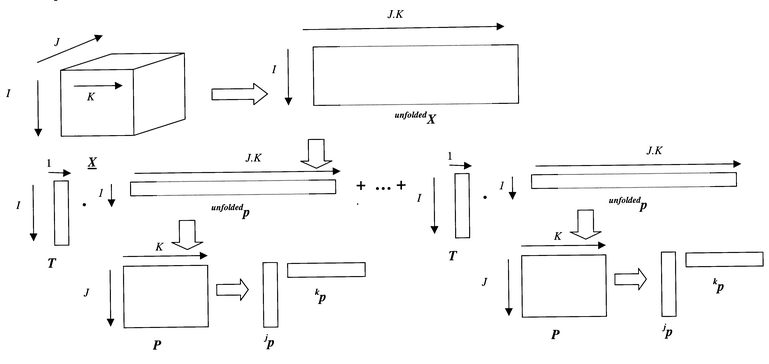
The experimental data of case study 2 can be considered to be arranged in the form of a cube, with three dimensions, I for the number of samples, and J and K for the measurements. For case study 2, there are: I = 14 samples; J = 40 sampling times in HPLC and K = 51 wavelengths.
Trilinear PLS1 attempts to model both the x and c blocks simultaneously. In this review we will illustrate the use with the algorithm of Appendix A2.4, based on methods proposed by de Jong36 and Bro.33
Superficially, trilinear PLS1 has many of the same objectives as normal PLS1, and the method is often represented diagrammatically as in Fig. 15, replacing ‘squares’ or matrices by ‘boxes’ or tensors, and replacing, where necessary, the dot product (‘.’) by something called a tensor product (‘⊗’). In fact, as we shall see, this is an oversimplification, and is not an entirely accurate description of the method.
 | ||
| Fig. 15 Principles of three way PLS. | ||
In trilinear PLS1, for each component it is possible to determine: a scores vector (t), of length I or 14 in this example; a weight vector, which has analogy to a loadings vector (jp) of length J or 40 in this example, referring to one of the dimensions (e.g. time), whose sum of squares equals 1, and another weight vector, which has analogy to a loadings vector (kp) of length K or 51 in this example, referring to the other one of the dimensions (e.g. wavelength) whose sum of squares also equals 1.
Superficially these vectors are related to scores and loadings in normal PLS, but in practice they are different, a key reason being that these vectors are not orthogonal in trilinear PLS1, influencing the additivity of successive components. In this paper, we keep the notation scores and loadings, simply for the purpose of compatibility with the rest of this article.
In addition, a vector q is determined after each new component,
by
| q = (T′.T)−1.T′. c |
| ĉ = T.q |
| c = T.q + f |
A key difference from bilinear PLS1 is that the elements of q have to be recalculated afresh as new components are computed, whereas for two-way PLS, the first element of q, is the same no matter how many components are calculated. This limitation is a consequence of non-orthogonality of components in the algorithms conventionally applied. Therefore, the concentration estimates are best expressed in matrix terms and not so easily as summations.
The x block residuals after each component are computed
conventionally by
| resid,axijk = resid,a −1x − tij pjk pk |
Sometimes these equations are written as tensor products, but there are a large number of ways of multiplying tensors together, so this notation can be confusing.

However, tensors are simply methods for rearranging the data, and it is often conceptually more convenient to deal directly with vectors and matrices, just as in Section 2.5.1 by unfolding the data. This procedure can be called matricisation.
In mathematical terms we can state that

| Pa = jpa .kp a |
Fig. 16 represents this procedure, avoiding tensor multiplication, using conventional matrices and vectors together with unfolding. A key problem with the common implementation of trilinear PLS1 is that, since the scores and loadings of successive components are not orthogonal, the methods for determining residuals in simply an approximation. Hence the x block residuals do not have a direct physical meaning. It also means that there are no obvious analogies to eigenvalues. This means that it is not easy to determine the size of the components or the modelling power using the x scores and loadings, but, nevertheless, the concentration (or c block) is modelled well. Since the prime aim of calibration is to predict concentrations rather than spectra or chromatograms, trilinear PLS1 is adequate, provided that care is taken to interpret the output.
 | ||
| Fig. 16 Three way calibration using unfolded matrix notation as discussed in Section 2.5.2. | ||
In order to understand this method further, a small simulated example is given in Table 17, consisting of a 4 × 5 × 6 array, originating from three compounds, whose concentrations are also presented. No noise is added, so that there should be an exact model after three PLS components. Trilinear PLS1 is performed on the first compound. The main results for the first compound are given in Table 18. It can be seen that three components provide an exact model of the concentration, but there is still an apparent residual error in the x matrix, representing 2.51% of the overall sum of squares of the data (=4.03 × 107): this error has no real physical or statistical meaning, except that it is small. Despite this, it is essential to recognise that the concentration has been modelled well, so for the purpose of calibration the algorithm has performed well.
| (a) X data | |||||
| 390 | 421 | 871 | 940 | 610 | 525 |
| 635 | 357 | 952 | 710 | 910 | 380 |
| 300 | 334 | 694 | 700 | 460 | 390 |
| 65 | 125 | 234 | 238 | 102 | 134 |
| 835 | 308 | 1003 | 630 | 1180 | 325 |
| 488 | 433 | 971 | 870 | 722 | 479 |
| 1015 | 633 | 1682 | 928 | 1382 | 484 |
| 564 | 538 | 1234 | 804 | 772 | 434 |
| 269 | 317 | 708 | 364 | 342 | 194 |
| 1041 | 380 | 1253 | 734 | 1460 | 375 |
| 186 | 276 | 540 | 546 | 288 | 306 |
| 420 | 396 | 930 | 498 | 552 | 264 |
| 328 | 396 | 860 | 552 | 440 | 300 |
| 228 | 264 | 594 | 294 | 288 | 156 |
| 222 | 120 | 330 | 216 | 312 | 114 |
| 205 | 231 | 479 | 481 | 314 | 268 |
| 400 | 282 | 713 | 427 | 548 | 226 |
| 240 | 264 | 576 | 424 | 336 | 232 |
| 120 | 150 | 327 | 189 | 156 | 102 |
| 385 | 153 | 482 | 298 | 542 | 154 |
| (b) c data (three components) | |||||
| 1 | 9 | 10 | |||
| 7 | 11 | 8 | |||
| 6 | 2 | 6 | |||
| 3 | 4 | 5 | |||
| t | jp | kp | q | ĉ | RMS concentration residuals | RMS of x “residuals” | |
|---|---|---|---|---|---|---|---|
| Component 1 | 3135.35 | 0.398 | 0.339 | 0.00140 | 4.38 | 20.79 | 2.35E+06 |
| 4427.31 | 0.601 | 0.253 | 6.19 | ||||
| 2194.17 | 0.461 | 0.624 | 3.07 | ||||
| 1930.02 | 0.250 | 0.405 | 2.70 | ||||
| 0.452 | 0.470 | ||||||
| 0.216 | |||||||
| Component 2 | −757.35 | −0.252 | 0.381 | 0.00177 | 1.65 | 1.33 | 1.41E+06 |
| −313.41 | 0.211 | 0.259 | 0.00513 | 6.21 | |||
| 511.73 | 0.392 | 0.692 | 6.50 | ||||
| −45.268 | 0.549 | 0.243 | 3.18 | ||||
| −0.661 | 0.485 | ||||||
| 0.119 | |||||||
| Component 3 | −480.37 | −0.875 | −0.070 | 0.00201 | 1 | 0.00 | 1.01E+06 |
| −107.11 | −0.073 | 0.263 | 0.00508 | 7 | |||
| −335.17 | −0.467 | 0.302 | 0.00305 | 6 | |||
| −215.76 | −0.087 | 0.789 | 3 | ||||
| 0.058 | 0.004 | ||||||
| 0.461 |
The application will be illustrated by the example of case study 2. The values of t, jp, kp and q for each successive component, are given in Table 19. The data are not centred. The predicted concentrations, formed by multiplying T and q, are given in Table 20. The first component contributes very little to the concentration estimate, most concentration estimates being extremely close to the average. This is because the impurity is in a small proportion and the data are uncentred, so the first component reflects primarily the overall chromatogram. At least three components are required for a sensible estimate of concentration.
| t1 | t2 | t3 | jp1 | jp2 | jp3 | kp1 | kp2 | kp3 | q1 | q2 | q3 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 9.424 | −0.450 | −0.195 | 0.000 | 0.001 | 0.001 | 0.180 | 0.164 | −0.152 | 0.00491 | 0.00012 | −0.00064 |
| 9.459 | −0.047 | 0.086 | 0.000 | 0.002 | 0.003 | 0.095 | 0.109 | −0.113 | 0.05116 | 0.09439 | |
| 9.643 | −0.211 | −0.100 | 0.000 | 0.005 | 0.008 | 0.039 | 0.075 | −0.091 | −0.21409 | ||
| 9.762 | −0.164 | −0.047 | 0.001 | 0.014 | 0.023 | 0.018 | 0.066 | −0.088 | |||
| 9.475 | −0.095 | −0.015 | 0.002 | 0.033 | 0.052 | 0.013 | 0.069 | −0.095 | |||
| 9.557 | −0.299 | −0.237 | 0.003 | 0.065 | 0.102 | 0.013 | 0.075 | −0.104 | |||
| 9.677 | 0.388 | 0.233 | 0.005 | 0.110 | 0.170 | 0.015 | 0.080 | −0.111 | |||
| 9.713 | −0.037 | −0.041 | 0.007 | 0.162 | 0.243 | 0.018 | 0.082 | −0.112 | |||
| 9.891 | 0.105 | 0.051 | 0.009 | 0.206 | 0.306 | 0.022 | 0.081 | −0.108 | |||
| 9.692 | 0.078 | −0.040 | 0.009 | 0.233 | 0.341 | 0.028 | 0.077 | −0.099 | |||
| 9.555 | 0.045 | −0.036 | 0.010 | 0.238 | 0.343 | 0.035 | 0.073 | −0.090 | |||
| 9.877 | 0.050 | −0.061 | 0.009 | 0.224 | 0.317 | 0.043 | 0.072 | −0.085 | |||
| 9.922 | 0.339 | 0.053 | 0.009 | 0.200 | 0.270 | 0.053 | 0.076 | −0.086 | |||
| 9.758 | −0.050 | −0.202 | 0.010 | 0.178 | 0.210 | 0.065 | 0.087 | −0.096 | |||
| 0.016 | 0.172 | 0.134 | 0.079 | 0.103 | −0.112 | ||||||
| 0.033 | 0.194 | 0.039 | 0.094 | 0.123 | −0.133 | ||||||
| 0.065 | 0.244 | −0.071 | 0.112 | 0.145 | −0.158 | ||||||
| 0.118 | 0.303 | −0.176 | 0.131 | 0.169 | −0.183 | ||||||
| 0.187 | 0.336 | −0.244 | 0.150 | 0.192 | −0.207 | ||||||
| 0.259 | 0.310 | −0.247 | 0.171 | 0.213 | −0.229 | ||||||
| 0.320 | 0.222 | −0.189 | 0.191 | 0.232 | −0.247 | ||||||
| 0.357 | 0.100 | −0.096 | 0.212 | 0.247 | −0.259 | ||||||
| 0.366 | −0.019 | −0.003 | 0.230 | 0.257 | −0.264 | ||||||
| 0.351 | −0.109 | 0.069 | 0.247 | 0.261 | −0.261 | ||||||
| 0.321 | −0.161 | 0.114 | 0.261 | 0.258 | −0.251 | ||||||
| 0.284 | −0.181 | 0.136 | 0.271 | 0.251 | −0.236 | ||||||
| 0.245 | −0.178 | 0.141 | 0.276 | 0.241 | −0.218 | ||||||
| 0.208 | −0.162 | 0.135 | 0.276 | 0.228 | −0.199 | ||||||
| 0.175 | −0.142 | 0.124 | 0.271 | 0.214 | −0.180 | ||||||
| 0.146 | −0.120 | 0.110 | 0.261 | 0.200 | −0.165 | ||||||
| 0.123 | −0.100 | 0.095 | 0.244 | 0.187 | −0.154 | ||||||
| 0.103 | −0.083 | 0.081 | 0.222 | 0.173 | −0.145 | ||||||
| 0.087 | −0.069 | 0.068 | 0.195 | 0.158 | −0.137 | ||||||
| 0.074 | −0.057 | 0.057 | 0.167 | 0.143 | −0.128 | ||||||
| 0.064 | −0.047 | 0.049 | 0.140 | 0.128 | −0.119 | ||||||
| 0.055 | −0.039 | 0.041 | 0.111 | 0.111 | −0.108 | ||||||
| 0.048 | −0.032 | 0.035 | 0.080 | 0.092 | −0.095 | ||||||
| 0.042 | −0.027 | 0.031 | 0.052 | 0.073 | −0.082 | ||||||
| 0.037 | −0.022 | 0.026 | 0.030 | 0.056 | −0.068 | ||||||
| 0.033 | −0.019 | 0.023 | 0.015 | 0.043 | −0.056 | ||||||
| 0.007 | 0.034 | −0.046 | |||||||||
| 0.004 | 0.026 | −0.037 | |||||||||
| 0.002 | 0.020 | −0.029 | |||||||||
| 0.001 | 0.015 | −0.021 | |||||||||
| 0.001 | 0.011 | −0.015 | |||||||||
| 0.001 | 0.007 | −0.010 | |||||||||
| 0.000 | 0.005 | −0.007 | |||||||||
| 0.000 | 0.003 | −0.004 | |||||||||
| 0.000 | 0.002 | −0.003 | |||||||||
| 0.000 | 0.001 | −0.002 | |||||||||
| 0.000 | 0.001 | −0.001 |
| One component | Two components | Three components | |
|---|---|---|---|
| 0.046 | 0.024 | 0.018 | |
| 0.046 | 0.045 | 0.016 | |
| 0.047 | 0.038 | 0.033 | |
| 0.048 | 0.041 | 0.029 | |
| 0.047 | 0.043 | 0.031 | |
| 0.047 | 0.033 | 0.049 | |
| 0.048 | 0.069 | 0.049 | |
| 0.048 | 0.047 | 0.046 | |
| 0.049 | 0.055 | 0.048 | |
| 0.048 | 0.053 | 0.062 | |
| 0.047 | 0.050 | 0.056 | |
| 0.049 | 0.052 | 0.064 | |
| 0.049 | 0.067 | 0.082 | |
| 0.048 | 0.047 | 0.079 | |
| Error (%) | 42.267 | 35.981 | 5.534 |
The residual sum of squares from the x block might, at first glance, suggest a very different story and are presented in Table 21. Note that the first component accounts for by far the largest sum of squares, but the concentration is modelled very poorly using only one component, hence the x residuals do not provide very good information about the number of PLS components required to model the data adequately. It is important, also to be careful when interpreting these numbers as they are not true eigenvalues, unlike for bilinear PLS1.
| 0 components | 1311.218 |
| 1 component | 1.265 |
| 2 components | 0.601 |
| 3 components | 0.377 |
A beauty of multimode methods is that the dimensions of c (or indeed X) can be changed, for example, a matrix C can be employed consisting of several different compounds, exactly as in PLS2, or even, a tensor. It is possible to define the number of dimensions in both the x and c blocks, for example, a three way x block and a two way c block may consist of a series of two-way chromatograms each containing several compounds. However, unless one has a good grip of the theory or there is a real need from the nature of the data, it is best to reduce the problem to one of trilinear PLS1: for example a concentration matrix C can be treated as several concentration vectors, in the same way that a calibration problem that might appear to need PLS2 can be reduced to several calculations using PLS1.
Whereas there has been a huge interest in multimode calibration in the theoretical chemometrics literature, there are important limitations to the applicability of such techniques. Good, very high order, data are rare in analytical chemistry. Even three-way calibration, such as in DAD-HPLC, has to be used cautiously as there are frequent experimental difficulties with exact alignments of chromatograms in addition to interpretation of the numerical results. However, there have been some significant successes in areas such as sensory research and psychometrics.
3 Model validation
Unquestionably one of the most important aspects of all calibration methods is model validation. Numerous questions need to be answered. 1. How many significant components are needed to characterise a dataset? 2. How well is an unknown predicted? 3. How representative are the data used to produce a model?It is possible to obtain as close a fit as desired using more and more PLS or PCA components, until the raw data are fitted exactly; however, the later components are unlikely to be physically meaningful. There is a large literature on how to decide what model to adopt which requires an appreciation of model validation, experimental design and how to measure errors. Most methods aim to guide the experimenter as to how many significant components to retain. The methods are illustrated below with reference to PLS1 for brevity, but similar principles apply to all calibration methods, including those obtained using MLR, PCR, PLS2 and trilinear PLS1.
3.1 Autoprediction
The simplest approach to determining the number of significant components is by measuring the autoprediction error. This is also called the root mean square error of calibration. Usually (but not exclusively) the error is calculated on the concentration data matrix (c), and we will restrict the discussion below to errors in concentration: importantly, similar equations can be obtained for the x data.As more components are calculated, the residual error reduces. There are
two ways of calculating this error,

Note that these errors can easily be converted to a percentage variance or mean square error as described in Sections 2.3 and 2.4.

The value of 2Ecal will always decline in value as more components are calculated, whereas that of 1Ecal has the possibility of increasing slightly in size although, in most well behaved cases, will also reduce with number of components. If the 1Ecal does increase against component number it is indicative that there may be problems with the data. The two autopredictive errors for acenaphthylene, case study 1 and dataset A are presented in Fig. 17, using PLS1.
 | ||
| Fig. 17 2Ecal (bottom line) and 1Ecal (top line) against component number for acenaphthylene, as discussion in Section 3.1, plotted on a logarithmic scale for clarity. | ||
The autopredictive error can be used to determine how many PLS components to use in the model, in a number of ways.
1. A standard cut-off percentage error can be used, for example, 1%. Once the error has reduced to this cut-off, ignore later PLS (or PCA) components.
2. Sometimes an independent measure of the noise level is possible. Once the error has declined to the noise level, ignore later PLS (or PCA) components.
3. Occasionally the error can reach a plateau. Take the PLS components up to this plateau.
By plotting the eigenvalues (or errors in modelling the x block), it is also possible to determine prediction errors for the x data block. However, the main aim of calibration is to predict concentrations rather than spectra, so this information, whereas useful, is less frequently employed in calibration.
Many chemometricians do not like autoprediction as it is always possible to fit data perfectly simply by increasing the number of terms (or components) in the model. There is, though, a difference between statistical and chemical thinking. A chemist might know (or have a good intuitive feel) for parameters such as noise levels, and, therefore, in some circumstances be able to interpret the autopredictive errors successfully.
3.2 Cross-validation
An important chemometric tool is called cross-validation. The basis of the method is that the predictive ability of a model formed on part of a dataset can be tested out by how well it predicts the remainder of the data.It is possible to determine a model using I − 1 (=24) samples leaving out one sample (i). How well does this model fit the original data? Below we describe the method when the data are centred, the most common approach.
The following steps are used:
1. Centre both the I
− 1 (=24 in this example)
concentrations and spectra but remember to calculate the means
i and
i, involving
removing sample i and subtracting these means from the original
data.
2. Perform PLS to give a loadings matrix P for the x data and a loadings vector q for the c data. Note that the loadings will differ according to which sample is removed from the analysis.
Predicting the concentration of an unknown sample is fairly straightforward.
1. Call the spectrum of sample i xi(a row vector).
2. Subtract the mean of the I
− 1 samples from this to
give xi−i where
i is the mean
spectrum excluding sample i.
3. Calculate ti =
(xi
−i).p
where p are the loadings obtained from the PLS model using
I
− 1 samples excluding sample i.
4. Then calculate
cvĉi =
ti.q +
i which is the estimated
concentration of sample i using the model based on the remaining
(I
− 1) (=24 samples), remembering to add on the mean of
these samples again.
Most methods of cross-validation then repeat the calculation leaving
another spectrum out, and so on, until the entire procedure has been
repeated I (= 25) times over. The root mean square of these errors
is then calculated, as follows

For acenaphthylene using PLS1, the cross-validated error is presented in Fig. 18. An immediate difference between autoprediction and cross-validation is evident. In the former case the data will always be better modelled as more components are employed in the calculation, so the error will always reduce (with occasional rare exceptions in the case of 1Ecal). However, cross-validated errors normally reach a minimum as the correct number of components are found and then increase afterwards. This is because later components really represent noise and not systematic information in the data.
 | ||
| Fig. 18 Ecv for acenaphthylene using PLS1 as described in Section 3.2. | ||
Cross-validation has two main purposes.
1. It can be employed as a method for determining how many components characterise the data. From Fig. 18, it appears that nine components are necessary to obtain an optimum model for acenaphthylene. This number will rarely equal the number of chemicals in the mixture, as spectral similarities will often reduce this, whereas impurities and noise may increase it.
2. It can be employed as a fairly realistic error estimate for predictive ability. The minimum cross-validated prediction error for acenaphthylene of 0.040 mg L−1 equals 33.69%. This compares with an autopredictive error of 0.014 mg L−1 or 11.64% using ten components and PLS1 which is a very over-optimistic estimate.
Many refinements to cross-validation have been proposed in the literature. It is possible to perform cross-validation on the x block to determine the optimum number of components instead of the c block. There are several alternative approaches to cross- validation, a common one involving leaving larger proportions of the data out (e.g. one tenth) at a time, valuable for very large datasets. Some statisticians also propose methods involving removing individual measurements rather than individual objects or spectra, but such approaches are less used in analytical chemistry. The ‘leave one sample out at a time’ method is a popular, easily implemented, and widespread approach. There tends to be a significant divide between statisticians who may use a number of different sophisticated methods for cross-validation and analytical chemists who are generally satisfied with a straightforward approach.
3.3 Independent test sets
A significant weakness of cross-validation, is that it depends on the design and scope of the original dataset used to form the model. This dataset is often called a ‘training’ set. Consider a situation in which a series of mixture spectra are recorded, but it happens that the concentrations of two compounds are correlated, so that the concentration of compound A is high when compound B likewise is high, and vice versa. A calibration model can be obtained from analytical data, which predicts both concentrations well. Even cross-validation might suggest the model is good. However, if asked to predict a spectrum where compound A is in a high concentration and compound B in a low concentration it is likely to give very poor results, as it has not been trained to cope with this new situation. Cross-validation is very useful for removing the influence of internal factors such as instrumental noise or dilution errors but cannot help very much if there are correlations in the concentration of compounds in the training set.In some cases there will inevitably be correlations in the concentration data, because it is not easy to find samples without this. An example is in many forms of environmental monitoring. Several compounds often arise from a single source. For example, PAHs are well known pollutants, so if one or two PAHs are present in high concentrations it is a fair bet that others will be too. There may be some correlations, for example, in the occurrence of compounds of different molecular weights if a homologous series occurs, e.g. as the by-product of a biological pathway, there may be an optimum chain length which is most abundant in samples from a certain source. It would be hard to find field samples in which the concentrations of all compounds vary randomly. Consider, for example, setting up a model of PAHs coming from rivers close to several specific sources of pollution. The model may behave well on this training set, but can it be safely used to predict the concentrations of PAHs in an unknown sample from a very different source? Another serious problem occurs in process control. Consider trying to set up a calibration model using NIR to determine the concentration of chemicals in a manufacturing process. If the factory is behaving well, the predictions may be good, but it is precisely to detect problems in the process that the calibration model is effective: is it possible to rely on the predictions if data have a completely different structure?
Instead of validating the predictions internally, it is possible to test
the predictions against an independent data set, often called a
‘test’ set. Computationally the procedure is similar to
cross-validation. For example, a model is obtained using I
samples, and then the predictions are calculated using an independent test
set of L samples, to give

In case study 1, we can use the data arising from Table 1 (dataset A) for the training set (see above), but test the predictions using the spectra obtained from concentrations in Table 2 (dataset B). In this case, each dataset has the same number of samples, but this is not at all a requirement. The graph of Etest for acenaphthylene is presented in Fig. 19 and shows similar trends to that of Ecv although the increase in error when a large number of components are calculated is not so extreme. The minimum error is 35.78%, only slightly higher than for cross-validation. Normally the minimum test set error is higher than that for cross-validation, but if the structure of the test set is encompassed in the training set, these two errors will be very similar.
 | ||
| Fig. 19 Etestfor acynaphthylene, using PLS1 as described in Section 3.3. | ||
If, however, we use dataset B as the training set and dataset A as the test set, a very different story emerges as shown in Fig. 20 for acenaphthylene. The autopredictive and cross-validation errors are very similar to those obtained for dataset A: the value of 1Ecal using ten PLS components is 8.71%, compared with 11.34% and the minimum value of Ecv is 27.93% compared with 33.69%. However, the test set behaves differently and exhibits a minimum error of 49.12%, well in excess of the cross-validation error. Furthermore, Etest is always higher than Ecvexcept when a very large number of components have been calculated. When the model of dataset A is used to predict dataset B, it is found that there is not such a significant difference between the two types of errors.
 | ||
| Fig. 20 Autopredictive, cross−validation and test errors for dataset B (acynaphthylene) and PLS1. | ||
Hence dataset A is a good test set because it not only predicts itself well but also dataset B, but the reverse is not true. This suggests that dataset A encompasses the features of dataset B, but not the reverse. The reason for this apparent dichotomy will be discussed in greater detail in Section 3.4, and it is important to recognise that cross-validation can sometimes give a misleading and over-optimistic answer. However, this depends in part on the practical aim of the analysis. If, for example, data of the form of A are unlikely ever to occur, it is safe to use the model obtained from B for future predictions. For example, if it is desired to determine the amount of vitamin C in orange juices from a specific region of Spain, it might be sufficient to develop a calibration method only on these juices. It could be expensive and time-consuming to find a more representative calibration set. Is it really necessary or practicable to develop a method to measure vitamin C in all conceivable orange juices or foodstuffs? The answer is no, so, in some circumstances, living within the limitations of the original dataset is entirely acceptable. If at some future date extra orange juice from a new region is to be analysed, the first step is to set up a dataset from this new source of information as a test set and so determine whether the new data fit into the structure of the existing database or whether the calibration method must be developed afresh. It is, though, very important to recognise the limitations of calibration models especially if they are to be applied to situations that are wider than those represented by the initial training sets.
There are a number of variations on the theme of test sets, one being simply to take a few samples from a large training set and assign them to a test set, for example, take five out of the 25 samples from case study 1 (dataset A) and assign them to a test set, using the remaining 20 samples for the training set. Alternatively, datasets A and B could be combined, and 40 out of the 50 used for determining the model, the remaining ten for independent testing.
3.4 Experimental designs
One of the major problems arises in designing an adequate training set. In some cases it is not possible to control this easily (for example when sampling in the field) but in other situations, such as preparing mixtures in the laboratory, good design is possible.Many brush aside the design of training sets, often using empirical or random approaches. Some chemometricians recommend huge training sets of several thousand samples so as to get a representative distribution, especially if there are known to be half a dozen or more significant components in a mixture. In large industrial calibration models such a procedure is often considered important for robust predictions. This approach, though, is expensive in time and resources, and rarely possible in routine laboratory studies. More seriously, many instrumental calibration models are unstable, so calibration on Monday might vary significantly from calibration on Tuesday, hence if calibrations are to be repeated at regular intervals, the number of spectra in the training set must be limited. Finally very ambitious calibrations can take months or even years to develop, by which time instruments and often the detection methods are replaced. It is always important to consider resources available and balance how robust a model is required, and how frequently the measurement system will change.
For the most effective calibration models, it is best to determine carefully the nature of the training set using rational experimental design prior to investing time in experimentation. Provided that the spectra are linearly additive, and there are no serious baseline problems or interactions, there are standard designs that can be employed to obtain training sets. In fact, the majority of chemometric techniques for regression and calibration assume linear additivity. In the case where this may not be so, either the experimental conditions can be modified (for example if the concentration of a compound is too high so the absorbance does not obey the Beer–Lambert law the solution is simply diluted) or various approaches for multilinear modelling are required.
In calibration it is normal to use several concentration levels to form a model. Hence two-level designs37 (often presented as fractional factorials) are inadequate and typically four or five concentration levels are required for each compound. Chemometric techniques are most useful for multicomponent mixtures. Consider an experiment carried out in a mixture of methanol and acetone. What happens if the concentrations of acetone and methanol in a training set are completely correlated? If the concentration of acetone increases so does that of methanol, and similarly with a decrease. Such an experimental arrangement is shown in Fig. 21. A more satisfactory design is given in Fig. 22, in which the two concentrations are completely uncorrelated or orthogonal. In the former design there is no way of knowing whether a change in spectral characteristic results from change in concentration of acetone or of methanol. If this feature is consciously built into the training set and expected in all future samples, there is no problem, but if a future sample arises with a high acetone and low methanol concentration, calibration software will give a wrong answer for the concentration of each component. This is potentially very serious especially when the result of chemometric analysis of spectral data is used to make decisions, such as the quality of a batch of pharmaceuticals, based on the concentration of each constituent as predicted by computational analysis of spectra. In the absence of any certain knowledge (for example that in all conceivable future samples the concentrations of acetone and methanol will be correlated), it is safest to design the training set so that the concentrations of as many compounds as possible in a calibration set are orthogonal. Note that this type of orthogonality is different from the orthogonality or similarity between the spectra of each compound. Many users of chemometric calibration have a background in spectroscopy rather than experimental design and confuse these two concepts.
 | ||
| Fig. 21 Two factor, correlated, design. | ||
 | ||
| Fig. 22 Two factor, uncorrelated, design. | ||
A guideline to designing a series of multicomponent mixtures for calibration is described below: more details are available elsewhere.38,39
1. Determine how many components in the mixture (=k) and the maximum and minimum concentration of each component. Remember that, if studied by spectroscopy, the overall absorbance when each component is at a maximum should be within the Beer–Lambert limit (about 1.2 A for safety).
2. Decide how many concentration levels are required for each compound (=l), typically four or five. Mutually orthogonal designs are only possible if the number of concentration levels is a prime number or a power of a prime number, meaning that they are possible for 3, 4, 5, 7, 8 and 9 levels but not 6 or 10 levels.
3. Decide on how many mixtures to produce. Designs exist involving N = mlp mixtures, where l equals the number of concentration levels, p is an integer at least equal to 2, and m an integer at least equal to 1. Setting both m and p at their minimum values, at least 25 experiments are required to study a mixture (of more than one component) at five concentration levels.
4. The maximum number of mutually orthogonal compound concentrations in a mixture design where m = 1 is four for a three-level design, five for a four-level design and 12 for a five- level design. We will discuss how to extend the number of mutually orthogonal concentrations below. Hence choose the design and number of levels with the number of compounds of interest in mind.
The method for setting up a calibration design will be illustrated by a five-level, eight compound, 25 experiment, mixture, to give the design in Table 22.
 |
1. The first step is to number the levels, typically from −2 (lowest) to +2 (highest), corresponding to coded concentrations, e.g. the 0.7–1.1 mM; note that the coding of the concentrations can be different for each compound in a mixture.
2. Next, choose a repeater level, recommended to be the middle level, 0. For between 7 and 12 factors, and a five-level design, it is essential that this is 0. The first experiment is at this level for all factors.
3. Third, select a cyclical permuter for the remaining (l − 1) levels. This relates each of the four levels as will be illustrated below; only certain cyclic generators can be used namely −2→−1→2→1→−2 and −2→1→2→−1→−2 which have the property that factors j and j + l + 1 are orthogonal. For less than seven factors, the nature of this generator is not relevant, so long as it includes all four levels. One such permuter is illustrated in Fig. 23, used in the example below.
 | ||
| Fig. 23 A possible permuter | ||
4. Finally, select a difference vector; this consists of l − 1 numbers from 0 to l − 2, arranged in a particular sequence. Only a very restricted set of such vectors are acceptable of which [0 2 3 1] is an example. The use of the difference vector will be described below.
5. Then generate the first column of the design consisting of l2 (= 25) levels in this case, each level corresponding to the concentration of the first compound in the mixture in each of 25 experiments.
(a) The first experiment is at the repeater level for each factor.
(b) The l − 1 (=4) experiments 2, 8, 14 and 20 are at the repeater level. In general the experiments 2, 2 + l + 1, 2 + 2 (l + 1) up to 2 + (l − 1) × (l + 1) are at this level. These divide the columns into ‘blocks’ of five (= l) experiments.
(c) Now determine the levels for the first block, from experiments 3 to 7 (or in general terms experiments 3 to 2 + l). Experiment 3 can be at any level apart from the repeater. In the example below, we use level −2. The key to the conditions for the next four experiments is the difference vector. The conditions for the 4th experiment are obtained from the difference vector and cyclic generator. The difference vector [0 2 3 1] implies that the second experiment of the block is 0 cyclical differences away from the 3rd experiment or −2 using the cyclic permuter of Fig. 23. The next number in the difference vector is 2, making the 5th experiment at level 2 which is two cyclic differences from −2 . Continuing, the 6th experiment is three cyclic differences from the 5tht experiment or at level −1, and the final experiment of the block is at level 2.
(d) For the second block (experiments 9–13), simply shift the first block by one cyclic difference using the permuter of Fig. 23 and continue until the last (or fourth) block is generated.
6. Then generate the next column of the design as follows:
(a) The concentration of the second compound for the first experiment is always at the repeater level.
(b) The concentration for the second experiment is at the same level as the third experiment of the previous column, up to the 24th [ or (l2 − 1)th] experiment.
(c) The final experiment is at the same level as the second experiment for the previous compound.
7. Finally, generate successive columns using step 6 above.
The development of the design is illustrated in Table 22. Such designs can also be denoted l-level partial factorial designs. Note that a full five-level factorial design for eight compounds would require 58 or 390625 experiments, so there has been a dramatic reduction in the number of experiments required.
There are a number of important features to note about the design in Table 22. 1. In each column there are an equal number of −2, −1, 0, +1 and +2 levels. 2. Each column is orthogonal to every other column, that is the correlation coefficient is 0. 3. A graph of the levels of any two factors against each other is given in Fig. 24(a) for each combination of factors except factors 1 and 7, and 2 and 8, which graph is given in Fig. 24(b). It can be seen that in most cases the levels of any two factors are distributed exactly as they would be for a full factorial design, which would require almost half a million experiments. The nature of the difference vector is crucial to this important property. Some compromise is required between factors differing by l + 1 (or 6) columns, such as factors 1 and 7. This is unavoidable unless more experiments are performed.
 | ||
| Fig. 24 Graph of levels of (a) factor 2 versus 1 and (b) factor 7 versus 1 for the data of Table 20. | ||
It is possible to expand the number of factors using a simple trick of
matrix algebra. If a matrix A is orthogonal, then the
matrix

Any design can be checked for orthogonality, simply by determining the correlation coefficients between the concentrations of the various compounds. If the correlations are 0, then the design is a good one, and will result in a training set that spans the possible mixture space fairly evenly, whereas if there are correlations, the training set is less representative. Table 23 presents the correlations for datasets A and B of case study 1. It can be seen that the first is orthogonal, whereas the second contains correlations. This structure allows us to interpret the results of Section 3.3. The model from the well designed dataset A both predicts itself well and also predicts dataset B. However, although dataset B is well predicted using itself as a model (cross-validation) it does not provide such good predictions for the more representative dataset A.
| (a) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Py | Ace | Anth | Acy | Chry | Benz | Fluora | Fluore | Nap | Phen | |
| Py | 1.00 | |||||||||
| Ace | 0.00 | 1.00 | ||||||||
| Anth | 0.00 | 0.00 | 1.00 | |||||||
| Acy | 0.00 | 0.00 | 0.00 | 1.00 | ||||||
| Chry | 0.00 | 0.00 | 0.00 | 0.00 | 1.00 | |||||
| Benz | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 1.00 | ||||
| Fluora | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 1.00 | |||
| Fluore | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 1.00 | ||
| Nap | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 1.00 | |
| Phen | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 1.00 |
| (b) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Py | Ace | Anth | Acy | Chry | Benz | Fluora | Fluore | Nap | Phen | |
| Py | 1 | |||||||||
| Ace | 0.34 | 1 | ||||||||
| Anth | 0.34 | 0.34 | 1 | |||||||
| Acy | 0.04 | 0.34 | 0.34 | 1 | ||||||
| Chry | −0.38 | 0.04 | 0.34 | 0.34 | 1 | |||||
| Benz | −0.38 | −0.38 | 0.04 | 0.34 | 0.34 | 1 | ||||
| Fluora | −0.9 | −0.38 | −0.38 | 0.04 | 0.34 | 0.34 | 1 | |||
| Fluore | −0.34 | −0.9 | −0.38 | −0.38 | 0.04 | 0.34 | 0.34 | 1 | ||
| Nap | −0.34 | −0.34 | −0.9 | −0.38 | −0.38 | 0.04 | 0.34 | 0.34 | 1 | |
| Phen | −0.04 | −0.34 | −0.34 | −0.9 | −0.38 | −0.38 | 0.04 | 0.34 | 0.34 | 1 |
It is always important to consider the issues of design and orthogonality of training sets, as this provides clear guidance as to when the model is likely to perform adequately, and so the scope of the calibration.
4 Conclusions and discussion
There are many topics omitted from this review, some of which are listed below.Data preprocessing is important in multivariate calibration. Indeed, the relationship between even basic procedures such as centring the columns is not always clear, most investigators following conventional methods, that have been developed for some popular application but are not always appropriately transferable. Variable selection and standardisation can have a significant influence on the performance of calibration models.
Non-linearities occur in some forms of spectroscopy, especially when the absorbance is high, and greater effort has been made to enhance the basic PLS method to include squared and other terms. However, the analytical chemist will probably prefer to improve the experimental method of acquiring data. Non-linear calibration is most valuable in other areas of chemistry, such as QSAR, where a purely additive linear model is not necessarily expected.
Outlier detection is of concern in certain areas of science. The aim is to spot samples that do not appear to conform to the structure of the training set used to determine the calibration model. If outlying samples are treated in the normal way, inaccurate concentrations may be predicted; this is a consequence of experimental design of the training set. In the case of field samples, it is not always possible to produce training sets with orthogonal designs, so only samples with a similar structure will result in sensible predictions.
Multiway methods can be extended far beyond trilinear PLS1, and there are many cases in chemistry where such approaches are appropriate. However, in the case of calibration of analytical signals to determine concentrations, trilinear PLS1 is adequate in the majority of situations.
Users of a specific software package can often be overwhelmed by statistical output, but it is important to recognise that certain types of diagnostics are only really useful for particular problems. The huge influence that NIR had on the first applications of multivariate calibration has meant that several software packages are oriented heavily to the user of NIR instruments. Although a major success story in the history of chemometrics, it is for the analytical chemist to judge whether NIR will play a huge role in the future, or whether there are fertile new grounds for the detailed development of multivariate calibration methods throughout quantitative analytical chemistry.
It is important to understand the overall principles of the methods rather than rely too much on any individual piece of software or application. In fact the algorithms are straightforward and can be easily implemented computationally. For any individual instrumental technique, be it HPLC, or electrochemistry, or electronic absorption spectroscopy, and any specific application, such as process control or environmental monitoring, specific extensions are needed, and different workers from different scientific environments often assume that their own elaborations are generally transportable. This is often not the case, but a basic understanding of the methods reported in this paper provides a generic starting point for analytical calibration.
Acknowledgements
Steve Wilkes is thanked for obtaining case study 1 experimentally, and Kostas Zissis for case study 2. Les Erskine is thanked for producing Excel add-ins and validating the calculations, and Rasmus Bro for valuable comments on multiway calibration.Appendices
A1 Vectors and matrices
A vector consists of a row or column of numbers and is denoted as bold
lower case italics e.g.x. For example x = (7 8
11 −5) is a row vector and y =  a
column vector.
a
column vector.
A matrix is a two dimensional array of numbers and is denoted as bold
upper case italics e.g.X. For example X =
 is a matrix.
is a matrix.
The dimensions of a matrix are normally presented with the number of rows first and the number of columns second, and vectors can be represented as matrices with one dimension equal to 1, so that x above has dimensions 1 × 4 and X has dimensions 2 × 3.
A square matrix is one where the number of columns equals the number of
rows. For example  is a square matrix.
is a square matrix.
An identity matrix is a square matrix whose elements are equal to 1 in
the diagonal and 0 elsewhere, and is often called I. For example
I = is an identity matrix.
is an identity matrix.
The individual elements of a matrix are often referenced as scalars, with subscripts referring to the row and column; hence, in the matrix above, y21 = 6 which is the element in row 2 and column 1.
 then
then 
Matrix and vector multiplication using the ‘dot’ product is denoted by the symbol ‘.’ between matrices. It is only possible to multiply two matrices together if the number of columns of the first matrix equal the number of rows of the second matrix. The number of rows of the product will equal the number of rows of the first matrix, and the number of columns equal the number of columns of the second matrix. Hence a 3 × 2 matrix when multiplied by a 2 × 4 matrix will give a 3 × 4 matrix.
Multiplication of matrices is not commutative, that is A.B ≠ B.A even if the second product is allowable. Matrix multiplication can be expressed as summations. For arrays with more than two dimensions (e.g. tensors), conventional symbolism can be awkward and it is probably easier to think in terms of summations.
If matrix A has dimensions I × J and
matrix B has dimensions J × K then the
product C of dimensions I × K has elements
defined by


When several matrices are multiplied together it is normal to take any two neighbouring matrices, multiply them together and then multiply this product with another neighbouring matrix. It does not matter in what order this is done, hence A.B.C = (A.B).C = A.(B.C).
Most square matrices have inverses, defined by the matrix which when multiplied with the original matrix gives the identity matrix, and is represented by a −1 as a right-hand-side superscript, so that D.D−1 = I. Note that some square matrices do not have inverses: this is caused by there being correlations in the columns or rows of the original matrix.
An interesting property that chemometricians sometimes use is that the product of the transpose of a column vector with itself equals the sum of square of elements of the vector, so that x′.x = Σx2.
A2 Algorithms
There are many different descriptions of the various algorithms in the literature. This appendix describes one algorithm for each of four regression methods.Initialisation
1. Take a matrix Zand if required preprocess (e.g. mean-centre or standardise) to give the matrix X which is used for PCA.
The next principal component
2. Take a column of this matrix (often the column with greatest sum of
squares) as the first guess of the scores first principal component, call
it initial![[t with combining circumflex]](https://www.rsc.org/images/entities/b_i_char_0074_0302.gif) .
.
Iteration for each principal component
3. Calculate unicorm![[p with combining circumflex]](https://www.rsc.org/images/entities/b_i_char_0070_0302.gif) =
=
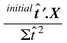 Comment: ideally t.p = X, so,
pre-multiplying each side by t′ we would
have t′.t. p =
∑t2p ≈
t′X for an exact fit, leading to the
approximation above.
Comment: ideally t.p = X, so,
pre-multiplying each side by t′ we would
have t′.t. p =
∑t2p ≈
t′X for an exact fit, leading to the
approximation above.
4. Normalise the guess of the loadings, so

5. Now calculate a new guess of the
scoresnew =
X.′ Comment: ideally t.p =
X, so, post-multiplying each side by p we would have t. p =
X.p′.p = X since p is
normalised, so its sum of squares equals 1.
Check for convergence
6. Check if this new guess differs from the first guess, a simple
approach is to look at the size of the sum of square difference in the old
and new scores, i.e.
Σ(initialt
−newt)2. If this is small the PC has been
extracted, set the PC scores and loadings for the current PC to
and . Otherwise return to step 3,
substituting the initial scores by the new scores.
Compute the component and calculate residuals
7. Subtract the effect of the new PC from the data matrix to get a residual data matrix residX = X − t.p.
Further PCs
8. If it is desired to compute further PCs, substitute the residual data matrix for X and go to step 2.
Initialisation
1. Take a matrix Z and if required preprocess (e.g. mean-centre or standardise) to give the matrix X which is used for PLS.
2. Take the concentration vector k and preprocess it to give the vector c which is used for PLS. Note that if the data matrix Z is centred down the columns, the concentration vector must also be centred. Generally, centring is the only form of preprocessing useful for PLS1. Start with an estimate of ĉ that is a vector of 0s (equal to the mean concentration if the vector is already centred).
The next PLS component
3. Calculate the vector h =
X′.c Comment: sometimes a weighting
vector s is employed, the aim being to obtain X from
c or the concentrations from the observed data. For a one
component mixture, ideally,
| X = c. s |
| X′ .X . s′ = X′ . c. s . s′ |
| X′ .X . s′ = X′ . c. s . s′ |

4. Calculate the scores which are simply given by

5. Calculate the x loadings by

6. Calculate the c loading (a scalar) by

Comment: note this calculation is identical in nature with the x loadings except that X is replaced by c.
Compute the component and calculate residuals
7. Subtract the effect of the new PC from the data matrix to get a
residual data matrix
| residX = X − t.p |
8. Determine the new concentration estimate by
| newĉ = initialĉ + t.q |
| residc = truec − ĉ |
Further PLS components
9. If further components are required, replace both X and c by the residuals and return to step 3.
Initialisation
1. Take a matrix Z and if required preprocess (e.g. mean-centre or standardise) to give the matrix X which is used for PLS.
2. Take the concentration matrix K and preprocess it to give the matrix C which is used for PLS. Note that if the data matrix is centred down the columns, the concentration vector must also be centred. Generally, centring is the only form of preprocessing useful for PLS2. Start with an estimate of Ĉ that is a matrix of 0s (equal to the mean concentration if the matrix is already centred).
3. An extra step is required to identify a vector u which can be a guess (as in PCA), but it can be chosen as one of the columns in the initial preprocessed concentration matrix, C.
The next PLS component
4. Calculate the vector
| h = X′.u |
5. Calculate the guessed scores by

6. Calculate the guessed x loadings by

7. Calculate the c loadings (a vector rather than scalar in PLS2)
by

8. If this is the first iteration, remember the scores, and call them
initialt, then produce a new vector
u by

Check for convergence
9. If this is the second time round compare the new and old scores
vectors, for example, by looking at the size of the sum of square
difference in the old and new scores, i.e.
Σ(initialt
−
newt)2. If this is small the PC has
been adequately modelled, set the PC scores and both types of loadings for
the current component to , and , and
![[q with combining circumflex]](https://www.rsc.org/images/entities/b_i_char_0071_0302.gif) . Otherwise calculate a new value of u as in step
8 and return to step 4.
. Otherwise calculate a new value of u as in step
8 and return to step 4.
Compute the component and calculate residuals
10. Subtract the effect of the new PC from the data matrix to get a
residual data matrix
| residX = X − t.p |
11. Determine the new concentration estimate by
| newĈ = Ĉ + t.q |
| residC = trueC − Ĉ |
Further PLS components
12. If further components are required, replace both X and C by the residuals and return to step 4.
Initialisation
1. Take a three way tensor Z and if required preprocess (e.g. mean-centre or standardise) to give the tensor X which is used for PLS. Perform all preprocessing on this tensor. The tensor has dimensions I × J × K. 2. Preprocess the concentrations if appropriate to give a vector c.
The next PLS component
3. From the original tensor, create a new matrix H with
dimensions J × K which is the sum of each of the I
matrices for each of the samples multiplied by the concentration of the
analyte for the relevant sample i.e.
| H = X1 c1 + X2 c2 + …. XIcI |
Comment: this is analogous to the vector h in PLS1, given by

4. Perform PCA on H to obtain the scores and loadings, gt and gp for the first PC of H. Note that only the first PC is retained, and for each PLS component a fresh H matrix is obtained. 5. Calculate the two x loadings for the current PC of the overall dataset by normalising the scores and loadings of Hi.e

Comment: in most cases hp will already be normalised, so the second step is not needed. 6. Calculate the overall scores by

Comment: this is sometimes expressed in the form of tensor multiplication, but this is not always an easy concept. However, there are strong analogies to PLS1, since

because the two loadings vectors are normalised, hence their sum of squares equals 1.Therefore, analogous to step 4 of PLS1

where the symbol ⊗ is sometimes used to indicate tensor multiplication. 7. Calculate the c loadings vector

| q= (T′.T)−1.T′.c |
Compute the component and calculate residuals
8. Subtract the effect of the new PC from the original data matrix to
get a residual data matrix (for each sample i)
| residX i = Xi − ti.j p.k p |
9. Determine the new concentration estimates by
| ĉ = T.q |
| residc = truec − ĉ |
Further PLS components
10. If further components are required, replace both X and c by the residuals and return to step 3.
References
- D. L. Massart, B. G. M. Vandeginste, S. N. Deming, Y. Michotte and L. Kaufman, Chemometrics: A Textbook, Elsevier, Amsterdam, 1988. Search PubMed.
- P. W. Araujo, D. A. Cirovic and R. G. Brereton, Analyst, 1996, 121, 581 RSC.
- C. Demir and R. G. Brereton, Analyst, 1998, 123, 181 RSC.
- K. D. Zissis, R. G. Brereton, S. Dunkerley and R. E. A. Escott, Anal. Chim. Acta, 1999, 384, 71 CrossRef CAS.
- D. A. Cirovic, R. G. Brereton, P. T. Walsh, J. Ellwood and E. Scobbie, Analyst, 1996, 121, 575 RSC.
- B. G. Osborne and T. Fearn, Near Infrared Spectroscopy in Food Analysis, Longman, London, 1986. Search PubMed.
- H. Martens and T. Næs, Multivariate Calibration, Wiley, Chichester, 1989. Search PubMed.
- P. Geladi and B. R. Kowalski, Anal. Chim. Acta, 1986, 185, 1 CrossRef CAS.
- S. Wold, H. Martens and H. Wold, Proc. Conf. Matrix Pencils, Lecture Notes in Mathematics, Springer-Verlag, Heidelberg, 1983, p. 286. Search PubMed.
- A. Høskuldsson, J. Chemom, 1988, 2, 211.
- R. Manne, Chemom. Intell. Lab. Syst., 1987, 2, 187 CrossRef CAS.
- P. J. Brown, J. R. Stat. Soc., Ser. B, 1982, 44, 287 Search PubMed.
- K. R. Beebe, R. J. Pell and M. B. Seasholtz, Chemometrics: A Practical Guide, Wiley, New York, 1998. Search PubMed.
- S. Wold, C. Albano, W. J. Dunn III, K. Esbensen, S. Hellberg, E. Johansson and M. Sjøstrøm, in Food Research and Data Analysis, ed. H. Martens and H. Russworm, Applied Science Publishers, London, 1983, p. 147. Search PubMed.
- http://www.infometrix.com/..
- http://www.camo.no/..
- http://www.umetrics.com/..
- http://www.eigenvector.com/..
- M. Thompson, D. W. Brown, T. Fearn, M. J. Gardner, E. J. Greenhow, R. Howarth, J. N. Miller, E. J. Newman, B. D. Ripley, K. J. Swan, A. Williams, R. Wood and J. J. Wilson, Analyst, 1994, 119, 2363 RSC.
- J. N. Miller, Analyst, 1991, 116, 3 RSC.
- J. N. Miller and J. C. Miller, Statistics for Analytical Chemistry, Ellis Horwood, Chichester, 1988. Search PubMed.
- C. Eisenhart, Ann. Math. Stat., 1939, 10, 162 Search PubMed.
- R. N. Draper and H. Smith, Applied Regression Analysis, Wiley, New York, 2nd edn., 1981. Search PubMed.
- D. L. Massart, B. G. M. Vandeginste, L. M. C. Buydens, S. de Jong, P. J. Lewi and J. Smeyers-Verbeke, Handbook of Chemometrics and Qualimetrics, Part A, Elsevier, Amsterdam, 1997. Search PubMed.
- R. J. Porra, in Chlorophyll, ed. H. Scheer, CRC Press, Boca Raton, FL, 1991, p. 31. Search PubMed.
- R. G. Brereton, Multivariate Pattern Recognition in Chemometrics, Illustrated by Case Studies, Elsevier, Amsterdam, 1992. Search PubMed.
- R. G. Brereton, Chemometrics: Applications of Mathematics and Statistics to Laboratory Systems, Ellis Horwood, Chichester, 1993. Search PubMed.
- S. Wold, K. Esbensen and P. Geladi, Chemom. Intell. Lab. Syst., 1987, 2, 37 CrossRef CAS.
- K. V. Mardia, J. T. Kent and J. Bibby, Multivariate Analysis, Academic Press, London, 1979. Search PubMed.
- R. G. Brereton, Analyst, 1995, 120, 2313 RSC.
- H. Martens and T. Næs, in Near-infrared Technology in Agricultural and Food Industries, ed. P. C. Williams and K. Norris, American Association of Cereal Chemists, St. Paul, MN, 1987, p. 57. Search PubMed.
- A. K. Smilde, P. Geladi and R. Bro, Multiway Analysis in Chemistry, Wiley, Chichester, 2000. Search PubMed.
- R. Bro, J. Chemom., 1996, 10, 47 CrossRef CAS.
- P. Geladi, Chemom. Intell. Lab. Syst., 1989, 7, 11 CrossRef CAS.
- S. Wold, P. Geladi, K. Esbensen and J. Öhman, J. Chemom., 1987, 1, 41 CAS.
- S. de Jong, J. Chemom., 1998, 12, 77 CrossRef CAS.
- S. N. Deming, S. L. Morgan, Experimental Design: A Chemometric Approach, Elsevier, Amsterdam, 2nd edn., 1993. Search PubMed.
- R. G. Brereton, Analyst, 1997, 122, 1521 RSC.
- J. Aybar Munoz and R. G. Brereton, Chemom. Intell. Lab. Syst., 1998, 43, 89 CrossRef.
Footnote |
| † Electronic Supplementary Information available. See http://www.rsc.org/suppdata/an/b0/b003805i/ |
| This journal is © The Royal Society of Chemistry 2000 |