A capillary electrophoresis microchip for the analysis of photographic developer solutions using indirect fluorescence detection
Somsak Sirichai and Andrew J. de Mello*
Zeneca/SmithKline Beecham Centre for Analytical Sciences, Department of Chemistry, Imperial College of Science, Technology and Medicine, London, UK SW7 2AY
First published on UnassignedUnassigned7th January 2000
Abstract
A capillary electrophoresis microchip is developed for the rapid determination of 4-amino-3-methyl- N-ethyl-N-(β-methane sulfonamidoethyl)aniline (CD-3) in commercial colour photographic processing solutions and the applicability of the method is examined. The use of indirect fluorescence as an on-chip detection method is also demonstrated. Using a running buffer at pH 11.9 prepared from disodium hydrogenphosphate and fluorescein the quantitative determination of CD-3 is achieved, resulting in an analysis time of approximately 7 s. Under these conditions, a detection limit of about 5 mg L−1 is obtained, with good linearity between signal and concentration over a range of 5–20 mg L−1.
Introduction
Miniaturisation of conventional analytical instrumentation has been the focus of much attention during the last decade. In particular, development of the concept of a miniaturized total analysis system (μ-TAS) has yielded specialised systems for genetic analysis, clinical diagnostics, chemical synthesis, drug screening, and environmental monitoring.1,2The advantages of ‘downsizing’ lie in improved efficiency with respect to sample size, response time, cost, throughput and automation. Ideally all steps of a complete analytical procedure (sample handling, chemical reactions, sample separation, detection and product isolation) should be performed on a single, integrated device. Normally, such devices are fabricated on glass or silicon substrates using standard micromachining methods (photolithography, etching, thin-film deposition and bonding). The result is a planar chip containing an enclosed channel manifold through which the sample can be manoeuvred. In addition, more complex components such as heaters, electrodes and mixers can be fabricated within the channel network.1,2
Miniaturisation of liquid phase separation methods has proved highly successful in planar chip formats, and a diversity have been successfully integrated into the concept of a μ-TAS. These include capillary electrophoresis (CE),3–8 free-flow electrophoresis (FFE),9,10 open-channel electrochromatography (CEC),11,12 open-channel liquid chromatography (LC),13,14 packed-bed chromatography,15 micellar electrokinetic capillary chromatography (MECC)16,17 and synchronised cyclic capillary electrophoresis (SCCE).18–20
The adaptation of conventional detection methods to measurement in small volumes has closely accompanied the development of μ-TAS. Indeed, it has long been realised that size limits for μ-TAS are primarily set by the system detector. Small volume detection in analytical systems has generally been based around optical measurements (either absorption or fluorescence). Unfortunately, small volume absorption measurements are compromised due to the difficulty in probing small volume cells, whilst maintaining a sufficiently long pathlength.21,22 With microfabricated devices this problem is exacerbated (due to reduced channel dimensions), and to date, fluorescence methods have proved far more useful. Detection limits for fluorescence based measurements are extremely low,23 and recently demonstrations of single molecule detection ‘on-chip’ have been reported.24 Although fluorescence techniques are inherently sensitive, they are costly and not applicable for all molecular systems (i.e., not all species that absorb radiation fluoresce).
Other approaches to on-chip detection have utilised electrochemiluminescence,25,26 electrochemical,27,28 and refractive index variation methods.29 In addition, CE microchips have been successfully coupled with electrospray mass spectrometry (ESMS).30 This approach extends the applicability of μ-TAS to molecules that are non-fluorescent, and leads to the possibility of high-throughput MS analysis in screening and diagnostic applications.
Another alternative for sensitive, universal detection is indirect fluorescence. This approach has been shown to be useful in visualising electrophoretic and chromatographic samples that would normally be impossible to detect without derivatisation.31–33 For capillary electrophoresis, a fluorescing anion is used as the buffer ion. A large fluorescence signal is therefore measured at the detector at all times. When analyte molecules pass into the detector volume, a lowering of the fluorescence background signal occurs, as solute ions displace fluorescent buffer ions (to maintain electrical neutrality in the zone).
This paper establishes the use of indirect fluorescence detection for the on-chip, electrophoretic analysis of a photographic colour developer, 4-amino-3-methyl-N-ethyl-N-(β-methane sulfonamidoethyl)aniline (CD-3), directly from a colour photographic processing solution.
Experimental
Microfabrication
All micromachining was performed in-house. Channels were fabricated using standard photolithographic procedures followed by wet chemical etching and bonding techniques. Briefly, a positive photoresist (S 1818, Shipley Corporation, Whitehall, PA, USA) was spin-coated onto the surface of a glass substrate (SLW, Hoya Corporation, Akishima-shi, Tokyo, Japan), and the channel design transferred to the substrate using a direct write laser lithography system (DWL2.0, Heidelberg Instruments, Heidelberg, Germany).34 After soft-baking (95 °C for 1 min) and exposure, the exposed regions of the photoresist were removed using a developer (Microposit 351, Shipley Europe Ltd, Coventry, UK) and the remaining photoresist hard-baked (95 °C for 5 min). Channels were then etched into the substrate using a buffered oxide etching solution (HF–NH4F) at ambient temperature. Once complete, the etched substrate was sonicated sequentially in acetone, H2SO4–H2O2 and ultra pure water at ambient temperature, and dried with N2 gas. Finally, a cover plate was thermally bonded to the substrate by heating the assembly at 550 °C for 1 h, 580 °C for 5 h and 555 °C for 1 h. The complete device was then allowed to cool for at least 8 h. Holes drilled in the top plate allow access to the fluidic network below.The microchip layout is shown in Fig. 1(a). All channels were 10 μm deep and 40 μm wide (the isotropic etching procedure results in a rounded channel profile, with a channel bed width of 20 μm). The channel connecting reservoirs 2 to 4 has a total capillary length of 3 cm and the channel connecting reservoirs 2 to 6 has a total capillary length of 10 cm. A double-T injector design6,35 was fabricated to allow injection of either 60 pL of sample (using a voltage between reservoirs 1 and 3) or 120 pL of sample (using a voltage between reservoirs 1 and 5).
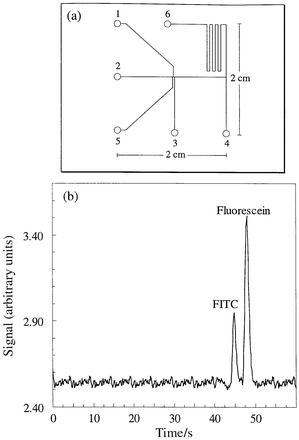 | ||
| Fig. 1 (a) Schematic diagram of the glass CE microchip used for all analyses. Reservoirs labelled 1–6: (1) sample outlet, (2) buffer solution inlet, (3) and (5) sample inlet, (4) and (6) outlet. (b) Electropherogram of a separation of fluorescein (100 μM) and fluorescein isothiocyanate (100 μM) on the planar glass microchip. The detection volume is located 4 cm downstream from the point of injection. Electric field = 300 V cm−1; total capillary length = 10 cm (between reservoirs 2 and 6). | ||
Instrumental
Electrophoretic separations were monitored on-chip via fluorescence and indirect fluorescence, using an inverted microscope (DMIL, Leica, Milton Keynes, UK) and filter cube (I3, Leica) that comprises an excitation filter (BP 450-490), a dichroic mirror (RKP 510), and a suppression filter (BP 515-560). Briefly, the excitation light from a 50 W mercury lamp (Leica) was passed through an excitation filter, reflected by the dichroic mirror and focused onto the microchip. The fluorescence emission was collected by a 10× microscope objective (0.42 NA, Newport, Irvine, CA, USA), passed through the dichroic mirror, a suppression filter, and a 20 μm diameter viewing window. A photomultiplier tube (MEA153, Seefelder Messtechnik, Germany) operating in current mode was used to detect sample emission. Data were acquired and stored as text files in a PC data acquisition program (PicoLog, Pico Technology, Hardwick, Cambridge, UK) and processed in Microsoft Excel 97.The in-house power supply used for electrophoresis was operated between 0 and +3 kV relative to ground and controlled by a programme written under the LabView 3.0 graphical programming environment (National Instruments, Austin, TX, USA). Before running experiments, the channels were flushed sequentially with NaOH, 18 MΩ Millipore water, and running buffer solution for 10 min, by applying vacuum to one reservoir and supplying the other five with the appropriate fluid. The microchip was operated in either a ‘sample loading’ or a ‘separation’ mode. To run a separation, platinum wires were inserted into the reservoirs (1 and 3 containing sample solution, and reservoirs 2 and 4 containing running buffer). All other reservoirs were filled with running buffer. In the ‘sample loading’ step, reservoir 1 is set to 0 V, reservoirs 2 and 4 are set to 3 kV, and reservoir 3 varied. In the ‘separation’ step, reservoirs 1 and 3 are set to 1.7 kV, reservoir 2 is set to 3 kV, and reservoir 4 is set at 0 V.
Chemicals
4-Amino-3-methyl-N-ethyl-N-(β-methane sulfonamidoethyl)aniline was a gift from Dr. T. Bumfrey (Kodak Ltd, Harrow, Middlesex, UK). Fluorescein di-sodium salt (Fluka, Gillingham, Dorset, UK) and disodium hydrogenphosphate (BDH-Merck, Poole, Dorset, UK) were used as received. All other chemicals were of analytical grade and prepared using high resistivity (18 MΩ), deionized water and filtered using 0.45 μm filters before introduction into the microchip. The pH of the running buffer containing fluorescein was adjusted by addition of NaOH.Results and discussion
Microstructure validation
Preliminary experiments, using conventional fluorescence detection, were performed to characterise the performance of the microchip for electrophoretic separations. Fig. 1(b) illustrates a typical free zone electrophoretic separation of fluorescein disodium salt and fluorescein isothiocyanate (FITC) performed on the glass microchip. As can be seen electrophoretic separation of the two dyes could be achieved within a few tens of seconds with satisfactory resolution using a separation electric field of 300 V cm−1. With a total capillary length of 10 cm the number of theoretical plates obtained was 16927 and 14127 for fluorescein disodium salt and FITC, respectively.Separation and detection of CD-3
As previously stated in indirect fluorescence detection, fluorescing ions in the running buffer create a constant fluorescence background. A signal is then obtained when fluorescing ions are displaced by non-fluorescent analyte ions. In this work, fluorescein (pK1 = 4.44 and pK2 = 6.36) was employed to generate a background signal. All experiments were performed in a phosphate buffer (pH 11.9) for a number of reasons. First, the fluorescence intensity of fluorescein is constant at pH![[greater than or equal, slant]](https://www.rsc.org/images/entities/char_2a7e.gif) 9,36 and secondly the pKa of CD-3
is 11.6.37 Furthermore, a pH of 11.9 was
chosen, since it represents a compromise between maintaining the buffer
capacity of the fluorescein solution and prolonging the life of the
microchip. Consequently, efficient charge displacement between CD-3 and
fluorescein anions should be feasible. Being a strong reducing agent, CD-3
is very sensitive to oxygen, especially in alkaline solution. The addition
of antioxidants is therefore essential in practice. A particularly good
choice is sulphite. The sulphite will react with oxygen in solution and
protect atmospheric oxygen; as a result, the oxidation of CD-3 is
reduced.38 For this reason, 0.1 M sulphite
was incorporated in all running buffers.
9,36 and secondly the pKa of CD-3
is 11.6.37 Furthermore, a pH of 11.9 was
chosen, since it represents a compromise between maintaining the buffer
capacity of the fluorescein solution and prolonging the life of the
microchip. Consequently, efficient charge displacement between CD-3 and
fluorescein anions should be feasible. Being a strong reducing agent, CD-3
is very sensitive to oxygen, especially in alkaline solution. The addition
of antioxidants is therefore essential in practice. A particularly good
choice is sulphite. The sulphite will react with oxygen in solution and
protect atmospheric oxygen; as a result, the oxidation of CD-3 is
reduced.38 For this reason, 0.1 M sulphite
was incorporated in all running buffers.By maintaining a constant concentration of phosphate in the running buffer, the influence of fluorescein concentration could be investigated. Fig. 2 shows the effect of fluorescein concentration on the analysis of a prepared CD-3 solution (1000 mg L−1). It can clearly be seen that both CD-3 and its oxidation product (OP) are detected within approximately five seconds of injection. Furthermore, as the concentration of fluorescein is increased, migration times increase (because of a decreased channel zeta potential that decreases the electroosmotic flow velocity) and resolution (bteween CD-3 and its OP) increases. According to this treatment, a fluorescein concentration of 2.0 mM was chosen for use in subsequent analyses of ‘real’ photographic process solutions.
 | ||
| Fig. 2 Electrophoretic analyses of CD-3 (1000 mg L−1) with varying amounts of fluorescein (0.2–2.0 mM in 10 mM phosphate buffer) in the running buffer. The detection volume is located 5 mm downstream from the point of injection. Electric field strength = 1 kV cm−1; total capillary length = 3 cm. | ||
The effect of buffer solution concentration on both peak signal and resolution are shown in Fig. 3(a) and 3(b). Resolution is calculated according to the following standard equation,39
 | (1) |
 | ||
| Fig. 3 (a) Effect of phosphate concentration on the signal of CD-3 and oxidation product peaks. (b) Effect of phosphate buffer concentration on the resolution between the CD-3 and oxidation product peaks. Electric field strength = 1 kV cm−1; total capillary length = 3 cm. | ||
Fig. 4(a) shows the influence of the injection voltage on the signal of CD-3. As expected, an increase in the applied voltage causes the peak signal to decrease. This reflects the fact that the volume of injected sample at the double-T injector decreases as the applied voltage increases. This effect was verified by injecting 20 mM fluorescein in 70 mM phosphate buffer into the system and visually observing fluorescence under the microscope. The relationship between injection time and signal was also examined and is described in Fig. 4(b). It can be seen that the intensity of the CD-3 peak dramatically increases in size as the injection time increases between 0 and 10 s. Above this value the signal intensity levels off. The results of both studies suggest an injection voltage of 2 kV and an injection time of 10 s for optimal analyses.
 | ||
| Fig. 4 (a) Effect of injection voltage on the peak signal of CD-3. The detection volume is located 5 mm downstream from the point of injection. (b) Effect of injection time on the signal of CD-3. Electric field strength = 1 kV cm−1; total capillary length = 3 cm; buffer composition = 2.0 mM fluorescein in 70 mM phosphate buffer (pH 11.9). | ||
Method validation
Using the optimal experimental parameters described above (a running buffer of 2 mM fluorescein in 70 mM phosphate, pH 11.9, and an injection voltage of 2 kV for 10 s) reproducibility, linearity and detection limit were studied. Run-to-run reproducibility on the microchip was very good. The standard deviation of the migration times for ten analyses of a standard solution of CD-3 was 0.54%. The linearity of the response was assessed using standard solutions of CD-3 (5–20 mg L−1). Under these conditions good linearity between signal and concentration was observed (r2 = 0.991). The detection limit (based on a minimum signal-to-noise ratio of 3) for the analysis of CD-3 in phosphate buffer was approximately 5 mg L−1, with a total analysis time of only 7.6 s.Analysis of RA-4 developer solutions
To establish the feasibility of using the microchip for on-line analysis of commercial photographic process solutions, a colour photographic developer solution from a RA-4 colour negative process (containing CD-3) was analysed using the methods described above. The only sample pre-treatment stage involves filtration of the sample through a 0.45 μm pore filter and dilution. Results of this analysis are illustrated in Fig. 5(a). It is immediately apparent that the sample matrix (developer additives) adversely affects background noise levels. Nevertheless sufficient sensitivity was achieved for successful analyte identification. Initial experiments utilising a separation length of 5 mm yielded a single peak (i.e., CD-3 and its OP could not be separated). Consequently, the separation length was increased to allow adequate discrimination of both molecular species. | ||
| Fig. 5 (a) Electrophoretic analysis of a RA-4 colour photographic developer solution. Electric field strength = 1 kV cm−1; total capillary length = 3 cm; buffer composition = 2.0 mM fluorescein in 70 mM phosphate buffer (pH 11.9). Electrokinetic injection at 2 kV for 10 s, (i) separation length = 5 mm, (ii) separation length = 9 mm, (iii) separation length = 13 mm. (b) Optimised electrophoretic analysis of RA-4 colour photographic developer solution. The detection volume is located 5 mm downstream from the point of injection. Electric field strength = 1 kV cm−1; total capillary length = 3 cm; buffer composition = 2.0 mM phosphate buffer (pH 11.9). Electrokinetic injection at 3 kV for 5 s. | ||
Since the sample matrix clearly affects both the efficiency and sensitivity of the analysis, new experimental parameters were optimised for the analysis of the commercial colour photographic developer solutions. Fig. 5(b) illustrates the results of the re-optimised analysis. In this case a running buffer of 2.0 mM fluorescein in 20 mM Na2HPO4 and 0.1 M Na2SO3 (pH 11.9), and an injection voltage of 3 kV for 5 s were used. Using these conditions an efficient separation of the developer and its oxidation product is obtained in approximately 7 s. The identity of the peaks is further confirmed by spiking the sample plug with a high concentration CD-3 solution. As expected, the longer time peak intensity increases.
Conclusions
The initial studies presented in this paper demonstrate the feasibility of using indirect fluorescence as a detection method for on-chip CE analysis. Furthermore, the successful determination of the reducing agent (developer) in commercial colour photographic process solutions, with minimal sample pre-treatment, demonstrates the possibility of using chip based CE systems for on-line process control applications. Detection limits achieved on the prototype chip, are sufficient for the determination of CD-3 in commercial processing solutions. Current studies are addressing improvements in detection limits and the integration of all component processes on the glass microchip (i.e. sample filtration and dilution). This should allow for the creation of a miniaturised analysis device that can periodically sample and analyse the composition of a colour photographic developer solution in situ.Acknowledgements
The authors would like to acknowledge Kodak Ltd and EPSRC UK for financial support. Furthermore the authors would like to extend special thanks to Mr. Gareth Jenkins for assistance with chip fabrication, Dr. Martin Kopp for building the high voltage power supply, and Dr. Trevor Bumfrey for the provision of photographic developers and useful advice. Finally, S.S. would like to thank the Royal Thai Government for provision of his research scholarship.References
- M. U. Kopp, H. J. Crabtree and A. Manz, Current Opin. Chem. Biol., 1997, 1, 410 Search PubMed.
- A. J. de Mello and A. Manz, in Microsystem Technology: A Powerful Tool for Biomolecular Studies, ed. J. M. Köhler, T. Mejevaia and H. P. Saluz, Birkhäuser, Switzerland, 1999, vol. 10, pp. 129–171. Search PubMed.
- A. T. Woolley and R. A. Mathies, Proc. Natl. Acad. Sci. USA, 1994, 91, 11348 CAS.
- J. D. Harrison, A. Manz, Z. Fan, H. Luedi and H. M. Widmer, Anal. Chem., 1992, 64, 1926 CrossRef.
- K. Seiler, D. J. Harrison and A. Manz, Anal. Chem., 1993, 65, 1481 CrossRef CAS.
- C. S. Effenhauser, A. Manz and H. M. Widmer, Anal. Chem., 1993, 65, 2637 CrossRef CAS.
- S. C. Jacobson, A. W. Moore and J. M. Ramsey, Anal. Chem., 1995, 67, 2059 CrossRef CAS.
- S. C. Jacobson, C. T. Culbertson, J. E. Daler and J. M. Ramsey, Anal. Chem., 1998, 70, 3476 CrossRef CAS.
- D. E. Raymond, A. Manz and H. M. Widmer, Anal. Chem., 1994, 66, 2858 CrossRef CAS.
- D. E. Raymond, A. Manz and H. M. Widmer, Anal. Chem., 1996, 68, 2515 CrossRef CAS.
- S. C. Jacobson, R. Hergenröder, L. B. Koutny and J. M. Ramsey, Anal. Chem., 1994, 66, 2369 CrossRef CAS.
- B. He, N. Tait and F. Regnier, Anal. Chem., 1998, 70, 3790 CrossRef CAS.
- A. Manz, Y. Miyahara, J. Miura, Y. Watanabe, H. Miyagi and K. Sato, Sens. Actuators, B, 1990, 1, 249 CrossRef.
- S. Cowen and D. H. Craston, Anal. Methods Instrum., 1996, Special Issue μ-TAS ’96, 197. Search PubMed.
- G. Ocvirk, E. Verpoorte, A. Manz, M. Grasserbauer and H. M. Widmer, Anal. Methods Instrum., 1995, 2, 74 Search PubMed.
- F. von Heeren, E. Verpoorte, A. Manz and W. Thormann, Anal. Chem., 1996, 68, 2044 CrossRef CAS.
- A. W. Moore, S. C. Jacobson and J. M. Ramsey, Anal. Chem., 1995, 67, 4184 CrossRef CAS.
- N. Burggraf, A. Manz, C. S. Effenhauser, E. Verpoorte, N. J. de Rooij and H. M. Widmer, J. High Resolut. Chromatogr., 1993, 16, 594 CAS.
- N. Burggraf, A. Manz, E. Verpoorte, C. S. Effenhauser and H. M. Widmer, Sens. Actuators, B, 1994, 20, 103 CrossRef CAS.
- F. von Heeren, E. Verpoorte, A. Manz and W. Thormann, J. Microcolumn, Sep., 1996, 8, 373 CrossRef CAS.
- E. Verpoorte, A. Manz, H. Lüdi, A. E. Bruno, F. Maystre, B. Krattiger, H. M. Widmer, B. H. van der Schoot and N. F. de Rooij, Sens. Actuators, B, 1992, 6, 66 CrossRef.
- Z. H. Lang, N. Chiem, G. Ocvirk, T. Tang, K. Fluri and D. J. Harrison, Anal. Chem., 1996, 68, 1040 CrossRef.
- M. D. Barnes, W. B. Whitten and J. M. Ramsey, Anal. Chem., 1995, 67, 418A CAS.
- J. C. Fister, S. J. Jacobson, L. M. Davis and J. M. Ramsey, Anal. Chem., 1998, 70, 431 CrossRef CAS.
- A. Arora, A. J. de Mello and A. Manz, Anal. Commun., 1997, 34, 393 RSC.
- G. C. Fiaccabrino, N. F. Rooij and M. Koudelka-Hep, Anal. Chim. Acta, 1998, 359, 263 CrossRef CAS.
- A. T. Woolley, K. Lao, A. N. Glazer and R. A. Mathies, Anal. Chem., 1998, 70, 684 CrossRef CAS.
- C. D. T. Bratten, P. H. Cobbold and J. M. Cooper, Anal. Chem., 1997, 69, 253 CrossRef CAS.
- N. Burggraf, B. Krattiger, A. J. de Mello, N. F. de Rooij and A. Manz, Analyst, 1998, 123, 1443 RSC.
- J. Li, P. Thibault, N. H. Bings, C. D. Skinner, C. Wang, C. Colyer and J. D. Harrison, Anal. Chem., 1999, 71, 3036 CrossRef CAS.
- W. G. Kuhr and E. S. Yeung, Anal. Chem., 1988, 60, 2642 CrossRef CAS.
- A. M. Desbene, C. J. Morin, N. L. Mofaddel and R. S. Groult, J. Chromatogr. A, 1995, 716, 279 CrossRef CAS.
- L. Gross and E. S. Yeung, Anal. Chem., 1990, 62, 427 CrossRef CAS.
- C. Schomburg, B. Höfflinger, R. Springer and R. Wijnaendts-van-Resandt, Microelectron. Eng., 1997, 35, 509 CrossRef CAS.
- D. J. Harrison, K. Fluri, K. Seiler, Z. Fan, C. S. Effenhauser and A. Manz, Science, 1993, 261, 895 CrossRef CAS.
- P. L. Desbene, C. J. Morin, A. M. D. Monvernay and R. S. Groult, J. Chromatogr. A., 1995, 689, 135 CrossRef CAS.
- T. H. James, The Theory of the photographic process, Macmillan Publishing Co., Inc., New York, 4th ed., 1977, pp. 317. Search PubMed.
- D. R. Canterford, Photogr. Sci. Eng., 1977, 21, 215 Search PubMed.
- L. S. Ettre, Pure Appl. Chem., 1993, 65, 819 CAS.
| This journal is © The Royal Society of Chemistry 2000 |