
Balancing charge-transfer strength and triplet states for deep-blue thermally activated delayed fluorescence with an unconventional electron rich dibenzothiophene acceptor†
 ‡b
Jonathan S.
Ward,
b
Andrew
Danos,
a
Andrei S.
Batsanov,
b
Martin R.
Bryce
*b
and
Fernando B.
Dias
*a
‡b
Jonathan S.
Ward,
b
Andrew
Danos,
a
Andrei S.
Batsanov,
b
Martin R.
Bryce
*b
and
Fernando B.
Dias
*a
Abstract
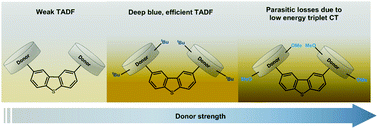
Manipulation of the emission properties of deep-blue emitters exhibiting thermally activated delayed fluorescence (TADF) through molecular design is challenging. We present an effective strategy to probe deeper into the role of localized (LE) and charge transfer (CT) states in the reverse intersystem crossing (RISC) mechanism. In a series of donor–acceptor–donor (D–A–D) blue emitters the dibenzothiophene functionality is used as an unconventional acceptor, while derivatives of 9,10-dihydro-9,9-dimethylacridine are used as electron-donors. tert-Butyl and methoxy substituents in the para-positions of the donor greatly enhance the donor strength, which allows exploration of different energy alignments among CT and LE triplet states. In the tert-butyl substituted compound the low energy triplet is localized on the acceptor unit, with the RISC mechanism (kRISC = 0.17 × 105 s−1) likely involving the mixture of CT and LE triplet states that are separated by less than 0.09 eV. An optimized organic light-emitting diode (OLED) based on the tBu-compound presents a maximum external quantum efficiency of 10.5% and deep-blue emission with Commission Internationale de l'Eclairage coordinates of (0.133, 0.129). However, when methoxy substituents are used, the low-energy triplet state moves away from the emissive 1CT singlet increasing the energy gap to 0.24 eV. Despite a larger ΔEST, a faster RISC rate (kRISC = 2.28 × 105 s−1) is observed due to the upper-state RISC occurring from the high-energy triplet state localized on the D (or A) units. This work shows the importance of fine-tuning the electronic interactions of the donor and acceptor units to control the TADF mechanism and achieve a deep-blue TADF OLED.
- This article is part of the themed collection: Journal of Materials Chemistry C Advisory Board Collection
 Please wait while we load your content...
Please wait while we load your content...

