
First-principles calculations on the intrinsic resistivity of borophene: anisotropy and temperature dependence
 *a
*a
Abstract
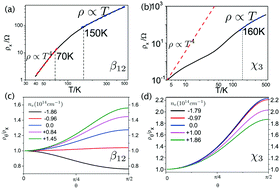
The intrinsic resistivity arising from electron–phonon scattering is an important property of metals, especially at room temperature. Borophene, a kind of successfully synthesized two-dimensional (2D) boron sheet, exhibits metallic electronic transport behavior. In this paper, we report first-principles calculations on the intrinsic resistivity of two borophene allotropes, called β12 and χ3. At first, we find that the intrinsic resistivity of the two structures shows non-trivial anisotropy which amounts to at least 28%. In addition, our calculations indicate that the intrinsic resistivity of β12 and χ3 are both proportional to temperature in the high-temperature region (>160 K). However, in the low-temperature region (<70 K), the T4-law of the intrinsic resistivity predicted by the Bloch–Grüneisen theory for 2D metals holds true for β12 but breaks down for χ3. Based on a detailed analysis, we find that the two borophenes have complicated Fermi surfaces with multiple branches. Optical phonon modes in β12 and the Umklapp process in χ3 play non-trivial roles in the low-temperature region. All of these factors influencing the intrinsic resistivity of borophene are beyond the simplifications adopted in the Bloch–Grüneisen theory. Therefore, we conclude that the Bloch–Grüneisen theory is inapplicable to explain the temperature dependence of the intrinsic resistivity of β12 and χ3, two experimentally available 2D metals.
 Please wait while we load your content...
Please wait while we load your content...

