
Exploring thermal transitions in anthradithiophene-based organic semiconductors to reveal structure-packing relationships†
 a
and
Chad
Risko
*a
a
and
Chad
Risko
*a
Abstract
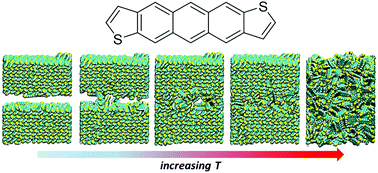
Thermal analyses provide macroscopic measures as to how molecular-scale features impact the solid-state packing of organic semiconductors. Here, we make use of molecular dynamics simulations to explore the phase transitions of a series of anthradithiophene-based molecular materials. Various models are explored to overcome superheating effects that are typically associated with the simulated annealing of crystalline materials using periodic boundary conditions. Slab models, in particular, are shown to provide good agreement with melt temperatures determined experimentally, especially for unsubstituted anthradithiophenes. Importantly, the simulations provide atomic-scale details regarding solid–solid and solid–liquid phase transitions that deliver key insights into how variations in the anthradithiophene chemistry impact the nature of the molecular packing and, in turn, can be used to enhance the rational engineering of crystal packing in these organic semiconductors.
 Please wait while we load your content...
Please wait while we load your content...

