
Optimizing thienothiophene chain lengths of D–π–D hole transport materials in perovskite solar cells for improving energy levels and hole mobility†
 *a
*a
Abstract
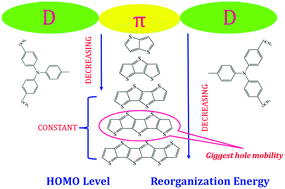
Although perovskite solar cells (PSCs) have recently achieved power conversion efficiencies (PCE) of over 23.6%, one major bottleneck for further improving the PCE is the lack of suitable hole transport materials. To further understand the structure–property relationship of hole transport materials and design new materials, we calculated the energy levels and optical properties of a series of thienothiophene derivatives by using density functional theory, and their hole transfer behaviors were also described by the Marcus charge transfer theory. It is found that the HOMO energies gradually decrease as the number of thiophene rings (n) increases when n is less than 4. However, when n is more than 4, the HOMO energy is a constant value of −5.23 eV. As for the LUMO energy and energy gaps, they show a similar change trend, that is, a gradual decrease with growing n. Optical calculations showed that thienothiophene extension cannot affect the Stokes shifts of thienothiophene derivatives. Importantly, it is found that the hole mobility of thienothiophene molecules is co-determined by the molecular size and odd or even number of thiophthene units, and all investigated thienothiophene molecules show higher hole mobility than Sprio-OMeTAD due to the face-to-face packing model. These results provide useful information to further develop suitable HTMs used in PSCs.
 Please wait while we load your content...
Please wait while we load your content...

