
Mechanism for spontaneous oxygen and hydrogen evolution reactions on CoO nanoparticles†
 ab
and
Alexie M.
Kolpak
*c
ab
and
Alexie M.
Kolpak
*c
Abstract
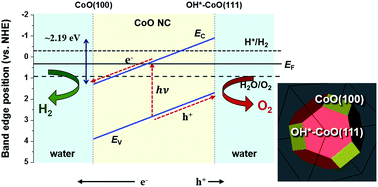
Overall photocatalytic water splitting with a high efficiency of ∼5% has recently been observed for CoO nanoparticle suspensions in the absence of an applied bias or co-catalyst. Although experimental measurements indicate that the overall photocatalytic water splitting is caused by optimal band edge alignments with respect to the redox potentials of water, the mechanism by which H2 and O2 simultaneously evolve on these nanoparticles is unknown. In this study, we used first-principles density functional theory (DFT) calculations to elucidate the mechanisms for the charge separation and H2 and O2 evolution on CoO nanoparticles under illumination in aqueous solution. We demonstrated that electrons are driven to the CoO(100) facet and holes are driven to the hydroxylated CoO(111) facet (OH*–CoO(111)) as a result of the built-in potential arising from the difference in the band edge positions on the two facets. Furthermore, based on a set of criteria, depending on if the photoexcited electrons and holes have sufficient energy to overcome the kinetic barrier along the H2 and O2 evolution reaction pathways, respectively, on the relevant surface facet, we show that H2 evolution preferentially occurs on the CoO(100) facet, while O2 evolution occurs on the OH*–CoO(111) surface. Our understanding of the overall water splitting mechanism on CoO nanoparticles provides a general explanation for the experimentally observed overall water splitting phenomena on a variety of self-standing photocatalysts, including γ-Ga2O3, Cu2O, and KTaO3, without an external driving potential or co-catalyst. In addition, we provide a new strategy for designing novel photocatalysts with high efficiency by controlling their surface configurations and morphologies.
- This article is part of the themed collection: 2019 Journal of Materials Chemistry A HOT Papers
 Please wait while we load your content...
Please wait while we load your content...

