
Unraveling the dominant phonon scattering mechanism in the thermoelectric compound ZrNiSn
 *a
and
*a
and
Abstract
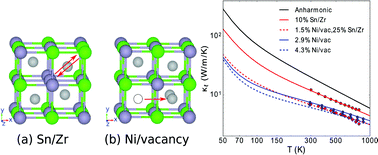
Determining defect types and concentrations remains a big challenge of semiconductor materials science. By using ab initio thermal conductivity calculations, we reveal that Ni/vacancy antisites, and not the previously claimed Sn/Zr antisites, are the dominant defects affecting thermal transport in the half-Heusler compound ZrNiSn. Our calculations correctly predict the thermal conductivity dependence on temperature and concentration, in quantitative agreement with the published experimental results. Furthermore, we find a characteristic proportionality between phonon–antisite scattering rates and the sixth power of phonon frequency, for which we provide an analytic derivation. These results suggest that thermal conductivity measurements in combination with ab initio calculations can be used to quantitatively assess defect types and concentrations in semiconductors.
 Please wait while we load your content...
Please wait while we load your content...

