
Realistic numerical simulations of dendrimer molecules
Abstract
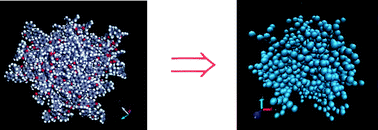
Dendrimer molecules have great potential in different applied fields because of their peculiar conformational properties. Many insights pertinent to the understanding of these properties can be provided by numerical simulation methods. Detailed models, based on atomistic representations of the molecular structures are usually employed for molecular dynamics simulations, which require an important amount of computational resources. More efficient Brownian dynamics or Monte Carlo methods can be more easily designed for coarse-grained models. However, the simplest models cannot distinguish between the different behaviours of particular dendrimer structures. Specific coarse-grained models that incorporate local details of the molecules structures are more adequate for these purposes. We describe some of the results provided by the simulation methods for particular dendrimer models and analyse their performance in the prediction of existing
 Please wait while we load your content...
Please wait while we load your content...

