
Porous aluminophosphates as adsorbents for the separation of CO2/CH4 and CH4/N2 mixtures – a Monte Carlo simulation study†

Abstract
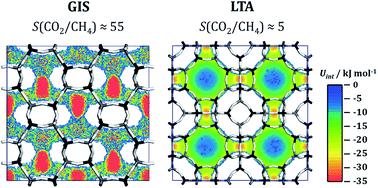
Aluminophosphates with zeolite-like topologies (AlPOs) have received considerable attention as potential adsorbents for use in the separation of methane-containing gas mixtures. Such separations, especially the removal of carbon dioxide and nitrogen from methane, are of great technological relevance in the context of the “upgrade” of natural gas, landfill gas, and biogas. While more than 50 zeolite frameworks have been synthesised in aluminophosphate composition or as heteroatom-substituted AlPO derivatives, only a few of them have been characterised experimentally with regard to their adsorption and separation behaviour. In order to predict the potential of a variety of AlPO frameworks for applications in CO2/CH4 and CH4/N2 separations, atomistic grand-canonical Monte Carlo (GCMC) simulations were performed for 53 different structures. Building on previous work, which studied CO2/N2 mixture adsorption in AlPOs (M. Fischer, Phys. Chem. Chem. Phys., 2017, 19, 22801–22812), force field parameters for methane adsorption in AlPOs were validated through a comparison to available experimental adsorption data. Afterwards, CO2/CH4 and CH4/N2 mixture isotherms were computed for all 53 frameworks for room temperature and total pressures up to 1000 kPa (10 bar), allowing the prediction of selectivities and working capacities for conditions that are relevant for pressure swing adsorption (PSA) and vacuum swing adsorption (VSA). For CO2/CH4 mixtures, the GIS, SIV, and ATT frameworks were found to have the highest selectivities and CO2 working capacities under VSA conditions, whereas several frameworks, among them AFY, KFI, AEI, and LTA, show higher working capacities under PSA conditions. For CH4/N2 mixtures, all frameworks are moderately selective for methane over nitrogen, with ATV exhibiting a significantly higher selectivity than all other frameworks. While some of the most promising topologies are either not available in pure-AlPO4 composition or collapse upon calcination, others can be synthesised and activated, rendering them interesting candidates for future experimental studies. In addition to predictions of mixture adsorption isotherms, further simulations were performed for four selected systems in order to investigate the microscopic origins of the macroscopic adsorption behaviour, e.g. with regard to the very high CH4/N2 selectivity of ATV and the loading-dependent evolution of the heat of CO2 adsorption and CO2/CH4 selectivity of AEI and GME.
 Please wait while we load your content...
Please wait while we load your content...

