
Combining machine learning and high-throughput experimentation to discover photocatalytically active organic molecules†

Abstract
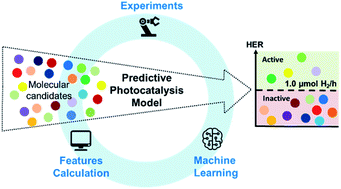
Light-absorbing organic molecules are useful components in photocatalysts, but it is difficult to formulate reliable structure–property design rules. More than 100 million unique chemical compounds are documented in the PubChem database, and a significant sub-set of these are π-conjugated, light-absorbing molecules that might in principle act as photocatalysts. Nature has used natural selection to evolve photosynthetic assemblies; by contrast, our ability to navigate the enormous potential search space of organic photocatalysts in the laboratory is limited. Here, we integrate experiment, computation, and machine learning to address this challenge. A library of 572 aromatic organic molecules was assembled with diverse compositions and structures, selected on the basis of availability in our laboratory, rather than more sophisticated criteria. This training library was then assessed experimentally for sacrificial photocatalytic hydrogen evolution using a high-throughput, automated method. Quantum chemical calculations and machine learning were used to visualise, interpret, and ultimately to predict the photocatalytic activities of these molecules, covering a much broader chemical space than for previous polymer photocatalyst libraries. By applying unsupervised learning to the molecular structures, we identified structural features that were common in molecules with high catalytic activity. Further analysis using calculated molecular descriptors within a suite of supervised classification algorithms revealed that light absorption, exciton electron affinity, electron affinity, exciton binding energy, and singlet–triplet energy gap had correlations with the photocatalytic performance. These trained predictive models can be used in future studies as filters to deprioritise or discard would-be low-activity candidate molecules from experiments, and to prioritize more favourable candidates. As a demonstration, we used virtual in silico experiments to show that it was possible to halve the experimental cost of finding 50% of the most active photocatalysts by using the machine learning model as an experimental advisor. We further showed that the ML advisor trained on the 572-molecule library could be used to make predictions for an unseen set of 96 molecules, achieving equivalent predictive accuracies to those in the initial training set. This marks a step toward the machine-learning assisted discovery of molecular organic photocatalysts and the approach might also be applied to problems beyond photocatalytic hydrogen evolution, such as CO2 reduction and photoredox chemistry.
- This article is part of the themed collections: Editor’s Choice: Malika Jeffries-EL and 2021 Chemical Science HOT Article Collection
 Please wait while we load your content...
Please wait while we load your content...

