
Method for accurate experimental determination of singlet and triplet exciton diffusion between thermally activated delayed fluorescence molecules†‡

Abstract
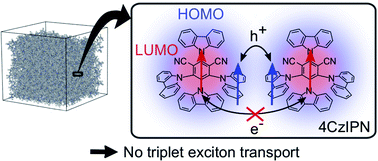
Understanding triplet exciton diffusion between organic thermally activated delayed fluorescence (TADF) molecules is a challenge due to the unique cycling between singlet and triplet states in these molecules. Although prompt emission quenching allows the singlet exciton diffusion properties to be determined, analogous analysis of the delayed emission quenching does not yield accurate estimations of the triplet diffusion length (because the diffusion of singlet excitons regenerated after reverse-intersystem crossing needs to be accounted for). Herein, we demonstrate how singlet and triplet diffusion lengths can be accurately determined from accessible experimental data, namely the integral prompt and delayed fluorescence. In the benchmark materials 4CzIPN and 4TCzBN, we show that the singlet diffusion lengths are (9.1 ± 0.2) and (12.8 ± 0.3) nm, whereas the triplet diffusion lengths are negligible, and certainly less than 1.0 and 1.2 nm, respectively. Theory confirms that the lack of overlap between the shielded lowest unoccupied molecular orbitals (LUMOs) hinders triplet motion between TADF chromophores in such molecular architectures. Although this cause for the suppression of triplet motion does not occur in molecular architectures that rely on electron resonance effects (e.g. DiKTa), we find that triplet diffusion is still negligible when such molecules are dispersed in a matrix material at a concentration sufficiently low to suppress aggregation. The novel and accurate method of understanding triplet diffusion in TADF molecules will allow accurate physical modeling of OLED emitter layers (especially those based on TADF donors and fluorescent acceptors).
 Please wait while we load your content...
Please wait while we load your content...

