
When are two hydrogen bonds better than one? Accurate first-principles models explain the balance of hydrogen bond donors and acceptors found in proteins†

Abstract
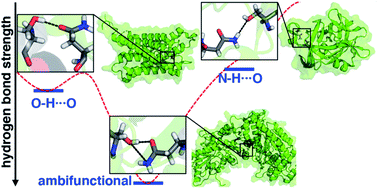
Hydrogen bonds (HBs) play an essential role in the structure and catalytic action of enzymes, but a complete understanding of HBs in proteins challenges the resolution of modern structural (i.e., X-ray diffraction) techniques and mandates computationally demanding electronic structure methods from correlated wavefunction theory for predictive accuracy. Numerous amino acid sidechains contain functional groups (e.g., hydroxyls in Ser/Thr or Tyr and amides in Asn/Gln) that can act as either HB acceptors or donors (HBA/HBD) and even form simultaneous, ambifunctional HB interactions. To understand the relative energetic benefit of each interaction, we characterize the potential energy surfaces of representative model systems with accurate coupled cluster theory calculations. To reveal the relationship of these energetics to the balance of these interactions in proteins, we curate a set of 4000 HBs, of which >500 are ambifunctional HBs, in high-resolution protein structures. We show that our model systems accurately predict the favored HB structural properties. Differences are apparent in HBA/HBD preference for aromatic Tyr versus aliphatic Ser/Thr hydroxyls because Tyr forms significantly stronger O–H⋯O HBs than N–H⋯O HBs in contrast to comparable strengths of the two for Ser/Thr. Despite this residue-specific distinction, all models of residue pairs indicate an energetic benefit for simultaneous HBA and HBD interactions in an ambifunctional HB. Although the stabilization is less than the additive maximum due both to geometric constraints and many-body electronic effects, a wide range of ambifunctional HB geometries are more favorable than any single HB interaction.
- This article is part of the themed collections: Welcoming our new Reaction Chemistry & Engineering Editorial Board members and Editor’s Choice – Graeme Day
 Please wait while we load your content...
Please wait while we load your content...

