
Mechanistic insights into rhodium-catalyzed enantioselective allylic alkylation for quaternary stereogenic centers†

Abstract
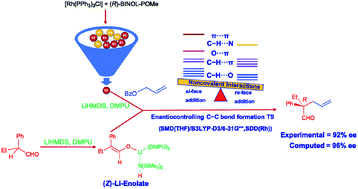
Installing quaternary stereogenic carbon is an arduous task of contemporary importance in the domain of asymmetric catalysis. To this end, an asymmetric allylic alkylation of α,α-disubstituted aldehydes by using allyl benzoate in the presence of Wilkinson's catalyst [Rh(Cl)(PPh3)3], (R)-BINOL–P(OMe) as the external ligand, and LiHMDS as the base has been reported to offer high enantioselectivity. The mechanistic details of this important reaction remain vague, which prompted us to undertake a detailed density functional theory (SMD(THF)/B3LYP-D3) investigation on the nature of the potential active catalyst, energetic features of the catalytic cycle, and the origin of high enantioselectivity. We note that a chloride displacement from the native Rh-phosphine [Rh(Cl)(PPh3)3] by BINOL–P(OMe) phosphite and an ensuing MeCl elimination can result in the in situ formation of a Rh-phosphonate [Rh(BINOL–P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)(PPh3)3]. A superior energetic span (δE) noted with such a Rh-phosphonate suggests that it is likely to serve as an active catalyst. The uptake of allyl benzoate by the active catalyst followed by the turnover determining C–O bond oxidative addition furnishes a Rh-π-allyl intermediate, which upon interception by (Z)-Li-enolate (derived from α,α-disubstituted aldehyde) in the enantiocontrolling C–C bond generates a quaternary stereogenic center. The addition of the re prochiral face of the (Z)-Li-enolate to the Rh-bound allyl moiety leading to the R enantiomer of the product is found to be 2.4 kcal mol−1 more preferred over the addition through its si face. The origin of the stereochemical preference for the re face addition is traced to improved noncovalent interactions (NCIs) and less distortion in the enantiocontrolling C–C bond formation transition state than that in the si face addition. Computed enantioselectivity (96%) is in very good agreement with the experimental value (92%), so is the overall activation barrier (δE of 17.1 kcal mol−1), which is in conformity with room temperature reaction conditions.
O)(PPh3)3]. A superior energetic span (δE) noted with such a Rh-phosphonate suggests that it is likely to serve as an active catalyst. The uptake of allyl benzoate by the active catalyst followed by the turnover determining C–O bond oxidative addition furnishes a Rh-π-allyl intermediate, which upon interception by (Z)-Li-enolate (derived from α,α-disubstituted aldehyde) in the enantiocontrolling C–C bond generates a quaternary stereogenic center. The addition of the re prochiral face of the (Z)-Li-enolate to the Rh-bound allyl moiety leading to the R enantiomer of the product is found to be 2.4 kcal mol−1 more preferred over the addition through its si face. The origin of the stereochemical preference for the re face addition is traced to improved noncovalent interactions (NCIs) and less distortion in the enantiocontrolling C–C bond formation transition state than that in the si face addition. Computed enantioselectivity (96%) is in very good agreement with the experimental value (92%), so is the overall activation barrier (δE of 17.1 kcal mol−1), which is in conformity with room temperature reaction conditions.
- This article is part of the themed collections: Celebrating the Chemical Sciences in India - Leaders in the Field Symposium 2021 and Celebrating the Chemical Sciences in India - Leaders in the Field Symposium 2020
 Please wait while we load your content...
Please wait while we load your content...

