
Using coligands to gain mechanistic insight into iridium complexes hyperpolarized with para-hydrogen†

Abstract
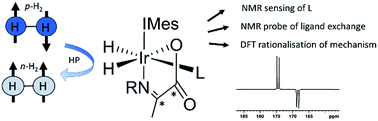
We report the formation of a series of novel [Ir(H)2(IMes)(α-13C2-carboxyimine)L] complexes in which the identity of the coligand L is varied. When examined with para-hydrogen, complexes in which L is benzylamine or phenethylamine show significant 1H hydride and 13C2 imine enhancements and may exist in 13C2 singlet spin order. Isotopic labeling techniques are used to double 13C2 enhancements (up to 750-fold) and singlet state lifetimes (up to 20 seconds) compared to those previously reported. Exchange spectroscopy and Density Functional Theory are used to investigate the stability and mechanism of rapid hydrogen exchange in these complexes, a process driven by dissociative coligand loss to form a key five coordinate intermediate. When L is pyridine or imidazole, competitive binding to such intermediates leads to novel complexes whose formation, kinetics, behaviour, structure, and hyperpolarization is investigated. The ratio of the observed PHIP enhancements were found to be affected not only by the hydrogen exchange rates but the identity of the coligands. This ligand reactivity is accompanied by decoherence of any 13C2 singlet order which can be preserved by isotopic labeling. Addition of a thiol coligand proved to yield a thiol oxidative addition product which is characterized by NMR and MS techniques. Significant 870-fold 13C enhancements of pyridine can be achieved using the Signal Amplification By Reversible Exchange (SABRE) process when α-carboxyimines are used to block active coordination sites. [Ir(H)2(IMes)(α-13C2-carboxyimine)L] therefore acts as unique sensors whose 1H hydride chemical shifts and corresponding hyperpolarization levels are indicative of the identity of a coligand and its binding strength.
- This article is part of the themed collection: 2019 Chemical Science HOT Article Collection
 Please wait while we load your content...
Please wait while we load your content...

