
Asymmetric synthesis of (−)-naltrexone†

Abstract
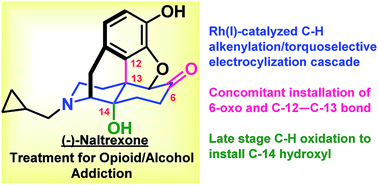
(−)-Naltrexone, an opioid antagonist used extensively for the management of drug abuse, is derived from naturally occurring opioids. Herein, we report the first asymmetric synthesis of (−)-naltrexone that does not proceed through thebaine. The synthesis starts with simple, achiral precursors with catalytic enantioselective Sharpless dihydroxylation employed to introduce the stereogenic centers. A Rh(I)-catalyzed C–H alkenylation and torquoselective electrocyclization cascade provides the hexahydro isoquinoline bicyclic framework that serves as the precursor to the morphinan core. The acidic conditions used for Grewe cyclization not only provide the morphinan framework, but also cause a hydride shift resulting in the introduction of the C-6 oxo functionality present in (−)-naltrexone. The C-14 hydroxyl group is installed by an efficient two-step sequence of Pd-mediated ketone to enone dehydrogenation followed by C–H allylic oxidation using Cu(II) and O2, a method that has not previously been reported either for the synthesis or semi-synthesis of opioids. The longest linear sequence is 17 steps, and because the stereogenic centers in the product rely on Sharpless asymmetric dihydroxylation, the route could be used to access either enantiomer of the natural product, which have disparate biological activities. The route also may be applicable to the preparation of opioid derivatives that could not be easily prepared from the more fully elaborated and densely functionalized opioid natural products that have traditionally served as the starting inputs.
- This article is part of the themed collection: Editor’s choice – Andrei Yudin
 Please wait while we load your content...
Please wait while we load your content...

