
The mechanism of a green fluorescent protein proton shuttle unveiled in the time-resolved frequency domain by excited state ab initio dynamics†
 ‡a
Nadia
Rega
*ab
‡a
Nadia
Rega
*ab
Abstract
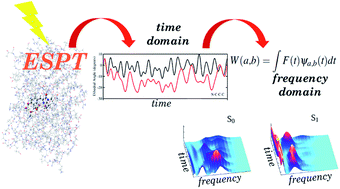
We simulated an excited state proton transfer in green fluorescent protein by excited state ab initio dynamics, and examined the reaction mechanism in both the time and the frequency domain through a multi resolution wavelet analysis. This original approach allowed us, for the first time, to directly compare the trends of photoactivated vibrations to femtosecond stimulated Raman spectroscopy results, and to give an unequivocal interpretation of the role played by low frequency modes in promoting the reaction. We could attribute the main driving force of the reaction to an important photoinduced softening of the ring–ring orientational motion of the chromophore, thus permitting the tightening of the hydrogen bond network and the opening of the reaction pathway. We also found that both the chromophore (in terms of its inter-ring dihedral angle and phenolic C–O and imidazolinone C–N bond distances) and its pocket (in terms of the inter-molecular oxygen’s dihedral angle of the chromophore pocket) relaxations are modulated by low frequency (about 120 cm−1) modes involving the oxygen atoms of the network. This is in agreement with the femtosecond Raman spectroscopy findings in the time-frequency domain. Moreover, the rate in proximity to the Franck Condon region involves a picosecond time scale, with a significant influence from fluctuations of nearby hydrogen bonded residues such as His148. This approach opens a new scenario with ab initio simulations as routinely used tools to understand photoreactivity and the results of advanced time resolved spectroscopy techniques.
- This article is part of the themed collection: Collection to celebrate our diverse and global authorship
 Please wait while we load your content...
Please wait while we load your content...

