
Quantitative first principles calculations of protein circular dichroism in the near-ultraviolet†

Abstract
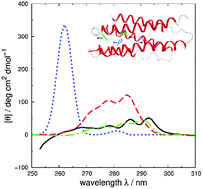
Vibrational structure in the near-UV circular dichroism (CD) spectra of proteins is an important source of information on protein conformation and can be exploited to study structure and folding. A fully quantitative theory of the relationship between protein conformation and optical spectroscopy would facilitate deeper interpretation of and insight into biophysical and simulation studies of protein dynamics and folding. We have developed new models of the aromatic side chain chromophores toluene, p-cresol and 3-methylindole, which incorporate ab initio calculations of the Franck–Condon effect into first principles calculations of CD using an exciton approach. The near-UV CD spectra of 40 proteins are calculated with the new parameter set and the correlation between the computed and the experimental intensity from 270 to 290 nm is much improved. The contribution of individual chromophores to the CD spectra has been calculated for several mutants and in many cases helps rationalize changes in their experimental spectra. Considering conformational flexibility by using families of NMR structures leads to further improvements for some proteins and illustrates an informative level of sensitivity to side chain conformation. In several cases, the near-UV CD calculations can distinguish the native protein structure from a set of computer-generated misfolded decoy structures.
 Please wait while we load your content...
Please wait while we load your content...

