
Manipulating charge transfer excited state relaxation and spin crossover in iron coordination complexes with ligand substitution†

Abstract
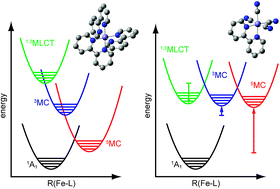
Developing light-harvesting and photocatalytic molecules made with iron could provide a cost effective, scalable, and environmentally benign path for solar energy conversion. To date these developments have been limited by the sub-picosecond metal-to-ligand charge transfer (MLCT) electronic excited state lifetime of iron based complexes due to spin crossover – the extremely fast intersystem crossing and internal conversion to high spin metal-centered excited states. We revitalize a 30 year old synthetic strategy for extending the MLCT excited state lifetimes of iron complexes by making mixed ligand iron complexes with four cyanide (CN−) ligands and one 2,2′-bipyridine (bpy) ligand. This enables MLCT excited state and metal-centered excited state energies to be manipulated with partial independence and provides a path to suppressing spin crossover. We have combined X-ray Free-Electron Laser (XFEL) Kβ hard X-ray fluorescence spectroscopy with femtosecond time-resolved UV-visible absorption spectroscopy to characterize the electronic excited state dynamics initiated by MLCT excitation of [Fe(CN)4(bpy)]2−. The two experimental techniques are highly complementary; the time-resolved UV-visible measurement probes allowed electronic transitions between valence states making it sensitive to ligand-centered electronic states such as MLCT states, whereas the Kβ fluorescence spectroscopy provides a sensitive measure of changes in the Fe spin state characteristic of metal-centered excited states. We conclude that the MLCT excited state of [Fe(CN)4(bpy)]2− decays with roughly a 20 ps lifetime without undergoing spin crossover, exceeding the MLCT excited state lifetime of [Fe(2,2′-bipyridine)3]2+ by more than two orders of magnitude.
- This article is part of the themed collection: Most downloaded articles of 2017: Inorganic and Physical Chemistry
 Please wait while we load your content...
Please wait while we load your content...

