
When the inhibitor tells more than the substrate: the cyanide-bound state of a carbon monoxide dehydrogenase†
Abstract
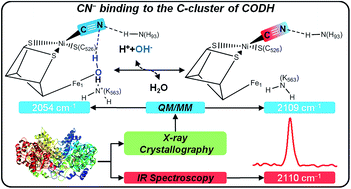
Carbon monoxide dehydrogenase (CODH) is a key enzyme for reversible CO interconversion. To elucidate structural and mechanistic details of CO binding at the CODH active site (C-cluster), cyanide is frequently used as an iso-electronic substitute and inhibitor. However, previous studies revealed conflicting results on the structure of the cyanide-bound complex and the mechanism of cyanide-inhibition. To address this issue in this work, we have employed IR spectroscopy, crystallography, site directed mutagenesis, and theoretical methods to analyse the cyanide complex of the CODH from Carboxydothermus hydrogenoformans (CODHIICh). IR spectroscopy demonstrates that a single cyanide binds to the Ni ion. Whereas the inhibitor could be partially removed at elevated temperature, irreversible degradation of the C-cluster occurred in the presence of an excess of cyanide on the long-minute time scale, eventually leading to the formation of [Fe(CN)6]4− and [Ni(CN)4]2− complexes. Theoretical calculations based on a new high-resolution structure of the cyanide-bound CODHIICh indicated that cyanide binding to the Ni ion occurs upon dissociation of the hydroxyl ligand from the Fe1 subsite of the C-cluster. The hydroxyl group is presumably protonated by Lys563 which, unlike to His93, does not form a hydrogen bond with the cyanide ligand. A stable deprotonated ε-amino group of Lys563 in the cyanide complex is consistent with the nearly unchanged C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching in the Lys563Ala variant of CODHIICh. These findings support the view that the proton channel connecting the solution phase with the active site displays a strict directionality, controlled by the oxidation state of the C-cluster.
N stretching in the Lys563Ala variant of CODHIICh. These findings support the view that the proton channel connecting the solution phase with the active site displays a strict directionality, controlled by the oxidation state of the C-cluster.
 Please wait while we load your content...
Please wait while we load your content...

