
Enantioselective synthesis of d-α-amino amides from aliphatic aldehydes†
Abstract
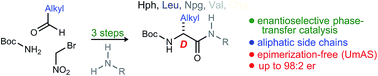
Peptides consisting of D-amino amides are highly represented among both biologically active natural products and non-natural small molecules used in therapeutic development. Chemical synthesis of D-amino amides most often involves approaches based on enzymatic resolution or fractional recrystallization of their diastereomeric amino acid salt precursors, techniques that produce an equal amount of the L-amino acid. Enantioselective synthesis, however, promises selective and general access to a specific α-amino amide, and may enable efficient peptide synthesis regardless of the availability of the corresponding α-amino acid. This report describes the use of a cinchona alkaloid-catalyzed aza-Henry reaction using bromonitromethane, and the integration of its product with umpolung amide synthesis. The result is a straightforward 3-step protocol beginning from aliphatic aldehydes that provides homologated peptides bearing an aliphatic side chain at the resulting D-α-amino amide.
 Please wait while we load your content...
Please wait while we load your content...

