
Computational prediction and experimental confirmation of B-site doping in YBa2Fe3O8†
Abstract
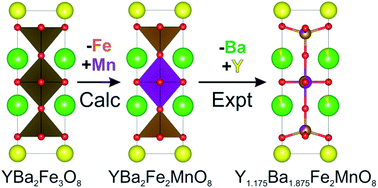
In this work we use calculations to obtain reaction enthalpies for the formation of YBa2Fe3−xMxO8 (where M = Co, Ni and Mn and x = 1, 2 and 3) from binary oxides and oxygen gas using Density Functional Theory (DFT). Based upon these calculations we are able to make predictions on favourable levels of doping and B-site ordering for YBa2Fe3−xMxO8, followed by experimental investigation in the same study. The composition where we predict doping to be favourable was experimentally investigated and a triple perovskite is found to be the major phase, confirming the prediction. Optimisation of the synthesis produced a phase-pure triple perovskite, Y1.175Ba1.825Fe2MnO8±δ, formed in a narrow compositional window. The crystal structure of this phase was analysed using Powder X-ray Diffraction (PXRD), iodometric titrations, Mössbauer spectroscopy and Neutron Powder Diffraction (NPD). This is the first reported example of ordered or disordered Fe and Mn coexistence in this structure type. We compare the observed structure against the initial DFT predictions and find them to be in good agreement and conclude that the computational methods presented within this work can be used as a predictive guide to the synthesis of oxide materials.
 Please wait while we load your content...
Please wait while we load your content...

