
Directional charge transfer and highly reducing and oxidizing excited states of new dirhodium(ii,ii) complexes: potential applications in solar energy conversion†
Abstract
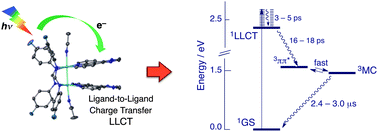
Two new series of dirhodium(II,II) complexes cis-[Rh2(μ-DTolF)2(L)2][BF4]2 and cis-[Rh2(μ-F-form)2(L)2][BF4]2 were synthesized and fully characterized (DTolF = p-ditolylformamidinate, F-form = p-difluorobenzylformamidinate; L = the chelating diimine ligands dpq (dipyrido[3,2-f:2′,3′-h]quinoxaline), dppz (dipyrido[3,2-a:2′,3′-c]phenazine) and dppn (benzo[i]dipyrido[3,2-a:2′,3′-h]quinoxaline). The complexes undergo facile oxidation and exhibit directed ligand-to-ligand charge transfer (LLCT) excited states upon excitation from the corresponding formamidinate to the diimine ligand. Time-resolved studies reveal that the LLCT states decay with lifetimes that range from 16 to 100 ps and generate a longer-lived excited state. For complexes with dpq and dppz ligands, the longer-lived excited state, with lifetimes that range from 40 to 100 ns, has been assigned as 3MC (MC = metal-centered) arising from the Rh2(π*) → Rh2(σ*) transition. In the case of cis-[Rh2(μ-DTolF)2(dppn)2]2+ and cis-[Rh2(μ-F-form)2(dppn)2]2+, the dppn-centered 3ππ* excited state is observed from ∼10 ps to ∼2 ns, but following its decay, the 3MC state of each complex is observed with lifetimes of 2.4 and 3.0 μs, respectively. The long lifetimes observed for the dppn complexes is explained by a pre-equilibrium of the low-lying 3ππ* and 3MC states. The excited state oxidation potentials, E*ox(1LLCT), for the complexes are calculated to lie between −2.5 and −2.8 V vs. SCE and E*ox(3MC) ∼ −1.8 V vs. SCE. The 1LLCT excited states of the complexes are also good oxidizing agents, with E*red(1LLCT) ∼ +1.3 V vs. SCE, making them significantly better oxidizing and reducing agents than commonly used Ru(II) complexes.
 Please wait while we load your content...
Please wait while we load your content...

