
First-principles prediction of chemically functionalized InN monolayers: electronic and optical properties

Abstract
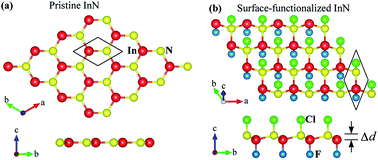
In this work, we consider the electronic and optical properties of chemically functionalized InN monolayers with F and Cl atoms (i.e., F–InN–F, F–InN–Cl, Cl–InN–F, Cl–InN–Cl monolayers) using first-principles calculations. The adsorption of the F and Cl atoms on the InN monolayer is determined to be chemically stable and the F–InN–F monolayer is most likely to occur. Our calculations show that the chemical functionalization with Cl and F atoms not only breaks the planar structure of InN monolayer but also increases its band gap. By using both Perdew, Burke, and Ernzerhof (PBE) and the Heyd–Scuseria–Ernzerhof (HSE06) hybrid functionals, all four models of chemically functionalized InN monolayers are found to be semiconductors with direct energy gaps and these gaps depend on the constituent species. When the spin–orbit coupling (SOC) was included, the energy gap of these monolayers was reduced and an energy splitting was found at the Γ-point in the valence band. Chemically functionalized InN monolayers can absorb light in a wide region, especially the F–InN–F and Cl–InN–F monolayers have a strong ability to absorb the visible light. Our findings reveal that the chemically functionalized InN monolayers have potential applications in next-generation optoelectronic devices.
 Please wait while we load your content...
Please wait while we load your content...

