
Computational study on the mechanism and kinetics for the reaction between HO2 and n-propyl peroxy radical†
 *a
Yanbo
Gai,
a
*a
Yanbo
Gai,
a
Abstract
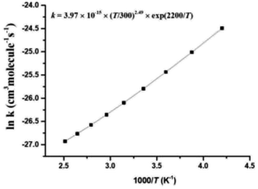
The n-propyl peroxy radical (n-C3H7O2) is the key intermediate during atmospheric oxidation of propane (C3H8) which plays an important role in the carbon and nitrogen cycles in the troposphere. In this paper, a comprehensive theoretical study on the reaction mechanism and kinetics of the reaction between HO2 and n-C3H7O2 was performed at the CCSD(T)/aug-cc-pVDZ//B3LYP/6-311G(d,p) level of theory. Computational results show that the HO2 + n-C3H7O2 reaction proceeds on both singlet and triplet potential energy surfaces (PESs). From an energetic point of view, the formation of C3H7O2H and 3O2 via triplet hydrogen abstraction is the most favorable channel while other product channels are negligible. In addition, the calculated rate constants for the title reaction over the temperature range of 238–398 K were calculated by the multiconformer transition state theory (MC-TST), and the calculated rate constants show a negative temperature dependence. The contributions of the other four reaction channels to the total rate constant are negligible.
 Please wait while we load your content...
Please wait while we load your content...

