
Theoretical calculation of a full-dimensional ab initio potential energy surface and prediction of infrared spectra for Xe–CS2
 †*ab
and
†*ab
and
Abstract
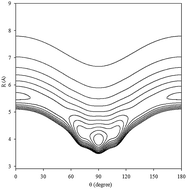
An effective four-dimensional (4D) ab initio potential energy surface (PES) for Xe–CS2 which explicitly involves the intramolecular Q1 symmetric stretching and Q3 antisymmetric stretching vibrational coordinates of CS2 is constructed. The computations are carried out employing single- and double-excitation coupled-cluster theory with a non-iterative perturbation treatment of triple excitations [CCSD(T)] method with a large basis set. Two vibrationally averaged potentials at the ground and ν1 + ν3 (ν1 = 1, ν3 = 1) excited states are obtained by integrating the 4D potentials over the Q1 and Q3 coordinates. The potentials have a T-shaped global minimum and two equivalent linear local minima. The radial discrete variable representation/angular finite basis representation and the Lanczos algorithm are employed to calculate the rovibrational energy levels for Xe–CS2. The infrared band origin shift associated with the fundamental band of CS2 is predicted, which is red-shifted by −1.996 cm−1 in the ν1 + ν3 region. In addition, we further predict the spectroscopic parameters for the ground and the ν1 + ν3 excited states of Xe–CS2. Compared with the previous Rg–CS2 (Rg = He, Ne, Ar, Kr) complexes, we found that the complexes of the rare gas atoms with CS2 display obvious regularities.
 Please wait while we load your content...
Please wait while we load your content...

