
Experimental and computational preference for phosphine regioselectivity and stereoselective tripodal rotation in HOs3(CO)8(PPh3)2(μ-1,2-N,C-η1,κ1-C7H4NS)†

Abstract
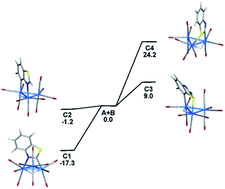
The site preference for ligand substitution in the benzothiazolate-bridged cluster HOs3(CO)10(μ-1,2-N,C-η1,κ1-C7H4NS) (1) has been investigated using PPh3. 1 reacts with PPh3 in the presence of Me3NO to afford the mono- and bisphosphine substituted clusters HOs3(CO)9(PPh3)(μ-1,2-N,C-η1,κ1-C7H4NS) (2) and HOs3(CO)8(PPh3)2(μ-1,2-N,C-η1,κ1-C7H4NS) (3), respectively. 2 exists as a pair of non-interconverting isomers where the PPh3 ligand is situated at one of the equatorial sites syn to the edge-bridging hydride that shares a common Os–Os bond with the metalated heterocycle. The solid-state structure of the major isomer establishes the PPh3 regiochemistry at the N-substituted osmium center. DFT calculations confirm the thermodynamic preference for this particular isomer relative to the minor isomer whose phosphine ligand is located at the adjacent C-metalated osmium center. 2 also reacts with PPh3 to give 3. The locus of the second substitution occurs at one of the two equatorial sites at the Os(CO)4 moiety in 2 and gives rise to a pair of fluxional stereoisomers where the new phosphine ligand is scrambled between the two equatorial sites at the Os(CO)3P moiety. The molecular structure of the major isomer has been determined by X-ray diffraction analysis and found to represent the lowest energy structure of the different stereoisomers computed for HOs3(CO)8(PPh3)2(μ-1,2-N,C-η1,κ1-C7H4NS). The fluxional behavior displayed by 3 has been examined by VT NMR spectroscopy, and DFT calculations provide evidence for stereoselective tripodal rotation at the Os(CO)3P moiety that serves to equilibrate the second phosphine between the two available equatorial sites.
 Please wait while we load your content...
Please wait while we load your content...

