
How the change of OMe substituent position affects the performance of spiro-OMeTAD in neutral and oxidized forms: theoretical approaches†
 *a
and
*a
and
Abstract
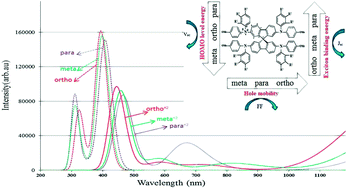
Density functional theory (DFT) was used to investigate the electronic and optical properties of the ortho, meta, and para derivatives of 2,2′,7,7′-tetrakis-(N,N-di-4-methoxyphenylamino)-9,9′spirobifluorene (spiro-OMeTAD) and its two oxidized forms (+1 and +2). The energy level, distribution shape, and density of highest occupied molecular orbital (HOMO) and of lowest unoccupied molecular orbital (LUMO) were computed for all three derivatives and compared in the neutral and oxidized forms. Results indicated that the different positions of OMe in the spiro-OMeTAD framework lead to different optical properties. It was also found that compared to the neutral form, in the oxidized forms, the maximum absorption band red shifts, new signals in the visible range between 500 and 850 nm appear, and the Stokes shift values reduce for all three derivatives. The exciton binding energy of spiro-OMeTAD with an o-OMe substituent is 0.52 eV, being smaller than that of p-OMe and m-OMe, indicating easier generation of free charge carriers. The hole mobility was calculated for all three molecules, and the obtained data revealed that the hole mobility of the o-OMe substituent has a value of 7.90 × 10−3 cm2 V−1 s−1, which is respectively 3 and 11 times larger than that of p-OMe and m-OMe. The smaller exciton binding energy and larger hole mobility of the o-OMe substituent will result in a higher short-circuit current density (Jsc) and a higher fill factor, respectively, demonstrating that po-spiro-OMeTAD is a promising candidate for use in perovskite solar cells. The reorganization energy, electron affinity, and ionization potential were also calculated and discussed.
 Please wait while we load your content...
Please wait while we load your content...

