
Computational prediction and experimental confirmation of rhombohedral structures in Bi1.5CdM1.5O7 (M = Nb, Ta) pyrochlores†

Abstract
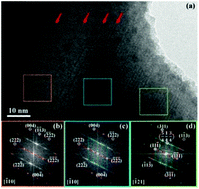
In this study, computationally predicted band gaps and structures using density functional theory (DFT) in Bi1.5CdM1.5O7 (M = Nb, Ta) pyrochlores are confirmed by experimental data on synthesized samples. Ordered Cd substitutions in the B-site of the pyrochlore structures are required to achieve electronic band gaps in the calculated energy band structures, when using full plane waves for DFT calculations, which are supported by a significantly lower total enthalpy. The computationally predicted band gap values are closely matched to experimental band gaps estimated from optical absorption spectra in the UV-Vis. In addition to the prediction of electronic structures, the models also indicate that the large ionic radius of the Cd-cation leads to symmetry modification from the archetypal cubic pyrochlore structure in Bi1.5CdM1.5O7 (M = Nb, Ta). A rhombohedral structure and localized superlattice order are confirmed using X-ray diffraction (XRD) and transmission electron microscopy (TEM) analysis. Energy dispersive X-ray spectroscopy profiles across the superlattice domain interfaces, which are constant within experimental uncertainty, indicate that domain formation is not compositionally driven but likely a mechanism to alleviate strain build up. Raman and FTIR spectroscopy analyses on these two compounds display strong similarities suggesting that peaks and activities belong to the same structure type.
 Please wait while we load your content...
Please wait while we load your content...

