
A computational study on interaction of aluminum with d-glucose 6-phosphate for various stoichiometries†
 *a
and
Xabier
Lopez
ab
*a
and
Xabier
Lopez
ab
Abstract
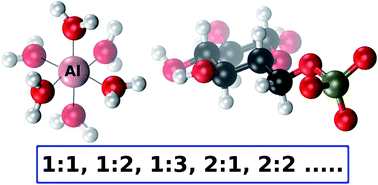
The interaction of aluminum with glucose 6-phosphate (Al-G6P) is thought to disrupt key processes of the glucide metabolism in cells. In this article, a Density Functional Theory study on the interaction of aluminum with D-glucose 6-phoshate is presented, combined with polarizable continuum models to account for bulk solvent effects. 143 aluminum–G6P complexes with different binding modes and various total charges are characterized comprising mononuclear (1 : 1, 1 : 2, 1 : 3, 1 : 1 : 1 (with citrate)) and dinuclear (2 : 1, 2 : 2) species. This large Al-G6P interaction dataset, the largest theoretical characterization of an aluminum–biophosphate interaction, gives insight into the diversity and complex picture of the interaction of aluminum with phosphate metabolites. We have found that charge and binding mode are driving factors in the binding affinity of glucose 6-phosphate. In addition, our calculations points to a tendency to form dicoordinated binding motifs, in which aluminum is bound to two functional groups of glucose 6-phosphate ligand. This tendency gives rise to a capacity of aluminum to act as a bridging agent in the coordination of several metabolites, a behavior that can be linked to the suspected tendency of aluminum to form aggregates that could induce various toxic effects in biological systems.
 Please wait while we load your content...
Please wait while we load your content...

