
Excited-state proton transfer in 4-2′-hydroxyphneylpyridine: full-dimensional surface-hopping dynamics simulations†
Abstract
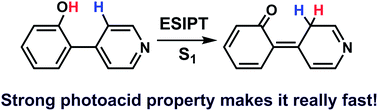
Herein we have employed combined electronic structure calculations (CASSCF and CASPT2) and “on-the-fly” fewest switches surface-hopping dynamics simulations (OM2/MRCI) to systematically study the S1 excited-state intramolecular proton transfer (ESIPT) and decay dynamics of 4-(2′-hydroxyphenyl)pyridine. On the basis of the optimized minima, conical intersections, and minimum-energy intramolecular proton transfer paths, we found that the S1 ESIPT process is essentially barrierless and results in a transient S1 keto species with a large Stokes shift. This keto species can be further decayed to the S0 state via the nearby keto S1/S0 conical intersection that can be easily approached structurally and energetically. In comparison, the other enol S1/S0 conical intersections are mechanistically less important. On the dynamical side, we have estimated the ESIPT is ultrafast and is complete within on average 80 fs. In addition, we have found that 94% trajectories hop to the S0 state via the keto S1/S0 conical intersection; while, the remaining 6% jump to the S0 state via the enol S1/S0 conical intersections. On arrival of the S0 state, the keto species will return to the enol one by an efficient ground-state reverse hydrogen transfer reaction.
 Please wait while we load your content...
Please wait while we load your content...

