
An efficient approach to explore the adsorption of benzene and phenol on nanostructured catalysts: a DFT analysis†
Abstract
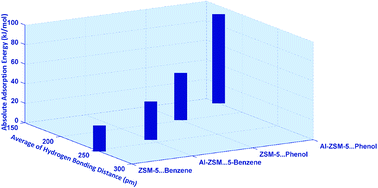
Adsorption of benzene and phenol on the 8T cluster model of ZSM-5 and Al-ZSM-5 catalysts, defined as ((H)3SiO)3–Si–O–Si–(OSi(H)3)3 and ((H)3SiO)3–Si–O(H)–Al–(OSi(H)3)3 structures, respectively, has been investigated comparatively using B3LYP, M06-2X, and wB97XD functionals employing the 6-311++G** standard basis set. Geometric parameters predict one and two types of hydrogen bonding in the guest-ZSM-5 and guest-Al-ZSM-5 complexes, respectively. Variations of adsorption energy, isotropic chemical shifts, δiso, of 1H, 17O, 27Al, and 29Si atoms contributing in the hydrogen bonding as well as quadrupole coupling constant, CQ, and asymmetry parameter, ηQ, of 2H, 17O, and 27Al atoms have been well correlated with the strength of the hydrogen bonds. Atom in molecules (AIM) calculations showed a covalent nature for the hydrogen bonds in the phenol⋯Al-ZSM-5 adsorption complex. Furthermore, based on AIM, NQR and NMR, the C–H⋯O and O–H⋯π hydrogen bonds have been confirmed in the benzene adsorbate zeolite, which may highlight a crucial feature of the adsorption of benzene molecules inside the pores of zeolites. The differences in the adsorption behavior between benzene and phenol on the ZSM-5 and Al-ZSM-5 are attributed to the differences in the strength of the hydrogen bonding interactions. Finally, Al-ZSM-5 appears to be an efficient adsorbent for phenol and benzene.
 Please wait while we load your content...
Please wait while we load your content...

