
A conceptual DFT study of the hydrogen trapping efficiency in metal functionalized BN system†
Abstract
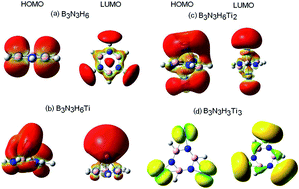
The hydrogen trapping efficiency of a metal functionalized BN system at various high electron density sites is studied using the first-principles conceptual density functional theory employing the M05-2X/6-311G+(d) level of theory. Metals are functionalized at three regions of the BN system, namely borazine, having high nucleus independent chemical shift values. H2 is trapped on the metal sites resulting in B3N3HXMiHm clusters [M = Li, Sc, Ti, V; X = 3, 6; i = 1–3; m up to 30]. Global reactivity attributes have been computed, which obey maximum hardness and minimum electrophilicity principles. The adsorption energy for physisorbed hydrogen is found to be low with Kubas–Niu interaction. Sc and Ti functionalized systems exhibit a combination of the hydrogen chemisorption and physisorption phenomena with storage capacity in the range 11.0–13.2 wt% hydrogen. These simple metal functionalized systems can be building blocks for assembly into two dimensional sheet or a multidecker complex, making it a potential hydrogen storage material.
 Please wait while we load your content...
Please wait while we load your content...

