
Investigation of the fracture mechanism of Cu–Al gradient structure
Abstract
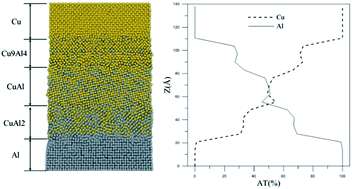
The mechanical properties of a Cu–Al gradient structure obtained from a tensile test have been investigated by molecular dynamics simulation. The tight binding potential was used to model the interaction between the Cu–Al atoms. The parameters of the TB potential were fitted according to the binding energies, atomic forces of the structures, and elastic constants of CuAl, CuAl2 and Cu3Al by density functional theory (DFT) calculations. These parameters were further applied to generate the Cu–Al gradient structure and perform the tensile test simulation. Meanwhile, the Honeycutt–Andersen (HA) index, radius distribution function and angular correlation function were used to analyze the characteristics of the atomic structure.
- This article is part of the themed collection: ChinaNANO
 Please wait while we load your content...
Please wait while we load your content...

