
Theoretical study of the reaction of hydroxyl radicals with uridine: the influence of ribose and solvent†
Abstract
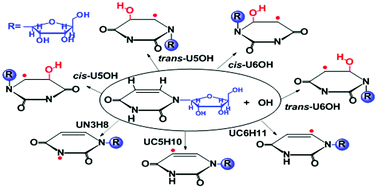
The reaction of hydroxyl radicals (˙OH) with uridine has been investigated using density functional theory (DFT). The influence of the environment was investigated using water and benzene as models for polar and non-polar surroundings, in addition to gas phase calculations. In order to imitate a much more realistic situation to that found in RNA, the ribose of the uracil ring was considered. Different pathways for the reaction of ˙OH with uridine were considered, involving addition and hydrogen abstraction reactions, with contributions to the overall reaction being around 99.9% and 0.1% at 298 K, respectively. The cis- and trans-U5OH addition reactions contributed around 62.2% and 35.6% in the polar surroundings, and about 70.1% and 26.8% in the non-polar surroundings, respectively, to the overall reaction. The cis-U5OH adduct was found to be the major product of the title reaction for all of the modeled environments. The steric effects of the ribose on the uracil ring makes U6OH addition more difficult than U5OH addition. Theoretical study of the reaction of ˙OH with uridine could help us to better understand oxidative damage to DNA and RNA. The good agreement found between the calculated data and the available experimental data supports the methodology used in this work, as well as the data reported here for the first time.
 Please wait while we load your content...
Please wait while we load your content...

